
Electrocatalysis
Ru oxides and substituted Ru oxides
click images to enlarge:
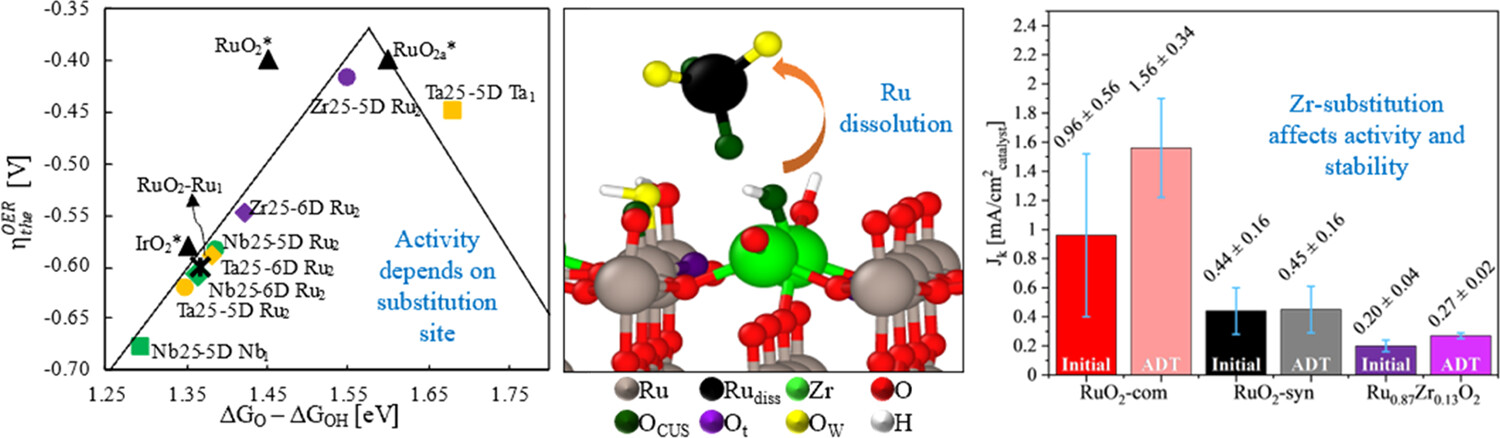
Francisco Ospina-Acevedo, Luis A. Albiter, Kathleen O. Bailey, José Fernando Godínez-Salomón, Christopher P. Rhodes, and Perla B. Balbuena, “Catalytic Activity and Electrochemical Stability of Ru1-xMxO2 (M = Zr, Nb, Ta): Computational and Experimental Study of the Oxygen Evolution Reaction,” ACS Appl. Materials & Interfaces, 2024, 16, 13, 16373–16398.
We use computations and experiments to determine the effect of substituting zirconium, niobium, and tantalum within rutile RuO2 on the structure, oxygen evolution reaction (OER) mechanism and activity, and electrochemical stability. Calculated electronic structures altered by Zr, Nb, and Ta show surface regions of electron density depletion and accumulation, along with anisotropic lattice parameter shifts dependent on the substitution site, substituent, and concentration. Consistent with theory, X-ray photoelectron spectroscopy experiments show shifts in binding energies of O-2s, O-2p, and Ru-4d peaks due to the substituents. Experimentally, the substituted materials showed the presence of two phases with a majority phase that contains the metal substituent within the rutile phase and a second, smaller-percentage RuO2 phase. Our experimental analysis of OER activity shows Zr, Nb, and Ta substituents at 12.5 atom % induce lower activity relative to RuO2, which agrees with computing the average of all sites; however, Zr and Ta substitution at specific sites yields higher theoretical OER activity than RuO2, with Zr substitution suggesting an alternative OER mechanism. Metal dissolution predictions show the involvement of cooperative interactions among multiple surface sites and the electrolyte. Zr substitution at specific sites increases activation barriers for Ru dissolution, however, with Zr surface dissolution rates comparable to those of Ru. Experimental OER stability analysis shows lower Ru dissolution from synthesized RuO2 and Zr-substituted RuO2 compared to commercial RuO2 and comparable amounts of Zr and Ru dissolved from Zr-substituted RuO2, aligned with our calculations.
Jose Fernando Godínez-Salomón, Francisco Ospina-Acevedo, Luis A. Albiter, Kathleen O. Bailey, Zachary G. Naymik, Rubén Mendoza-Cruz, Perla B. Balbuena, and Christopher P. Rhodes, “Titanium Substitution Effects on Structure, Activity, and Stability of Nanoscale Ruthenium Oxide Oxygen Evolution Electrocatalysts: Experimental and Computational Study,” ACS Appl. Nano Materials, 5, 8, 11752–11775, (2022).
Proton-exchange membrane water electrolyzers produce hydrogen from water and electricity and can be powered using renewable energy; however, the high overpotential, high cost, and limited supply of the oxygen evolution reaction (OER) electrocatalyst are key factors that hinder wide-scale adoption. Ruthenium oxide (RuO2) has a lower overpotential, lower cost, and higher global supply compared with iridium oxide (IrO2), but RuO2 is less stable than IrO2. As an approach to improve the catalytic stability, we report the effect of titanium substitution at different concentrations within nanoscale RuO2, Ru1–xTixO2 (x = 0–50 at. %), on the structure, OER activity, and stability using combined experiments and theory. Titanium substitution within rutile RuO2 affects the electronic structure, resulting in regions of electron accumulation and electron depletion at the surface, and shifts the d-band and O 2p band centers to higher binding energies. Calculations show that the effects of Ti on the electronic structure are highly dependent on not only the concentration but also the specific dopant location. From electrochemical testing and analysis of the electrolyte and simulations, titanium substitution at low concentrations (12.5 and 20 at. %) improves catalyst stability and lowers Ru dissolution. Experiments of OER activity agree with the theory that Ti substitution results in a higher overpotential when averaging over all adsorption sites. Theoretical analysis shows that specific sites predominately act as catalytic sites for the OER, while metal dissolution occurs at different sites. Specifically, OER has the lowest barriers at penta-coordinated Ru sites, while hexa-coordinated Ru sites have the lowest energetic barriers for dissolution.
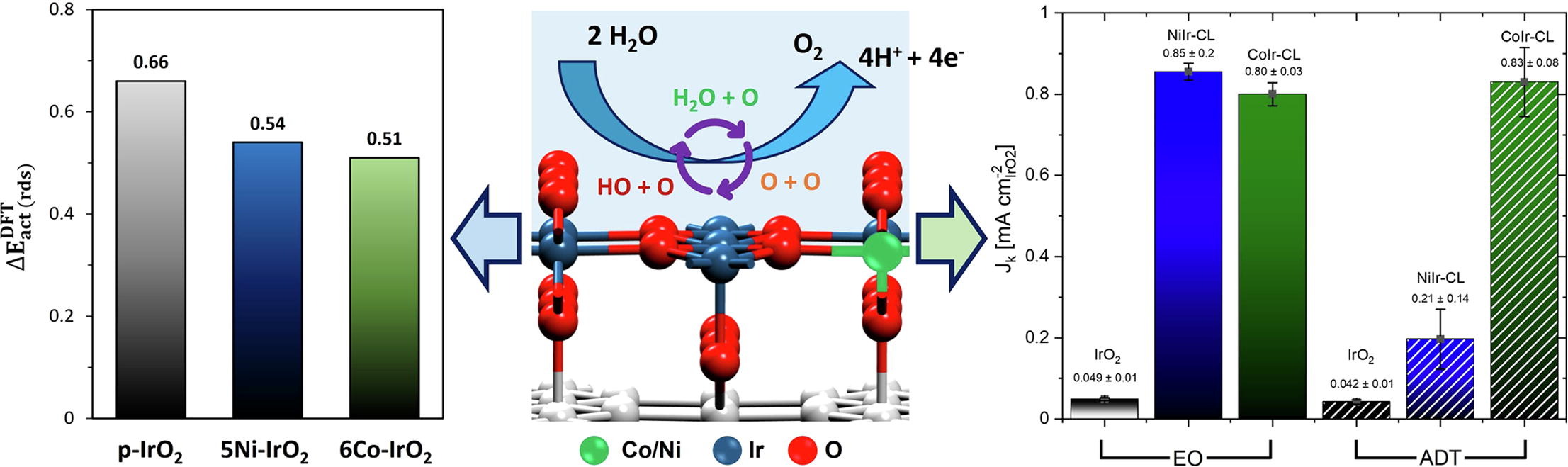
Luis E. Camacho-Forero, Fernando Godínez-Salomón, Guadalupe Ramos-Sánchez, Christopher P. Rhodes, and Perla B. Balbuena, “Theoretical and Experimental Study of the Effects of Cobalt and Nickel Doping within IrO2 on the Acidic Oxygen Evolution Reaction,” J. of Catalysis, 408, 64-80, (2022).
The effect of Ni and Co doping within IrO2 on the structure and oxygen evolution reaction (OER) was studied using integrated theory and experiments. Density Functional Theory (DFT) calculations show that the metal dopant influences the distribution of electronic charge and affects the thermodynamics and kinetics aspects of the OER when compared with undoped IrO2. Using DFT, multiple different reaction pathways were evaluated for O-O bond formation, and analysis supports the associative mechanism to be the most likely reaction pathway. Calculations showed lower activation energies for Ni and Co-doped IrO2 compared to undoped IrO2, in agreement with experimental analysis of the activation energy and OER activity. From experiments, Co-doping shows significantly improved stability compared to Ni-doping. Evaluation of the rate-determining step (rds) from calculations and experimental analysis shows apparent differences that indicate the additional factors need to be considered to enable improved correlation between theory and experiment regarding the OER rds.
Ru oxides and substituted Ru oxides
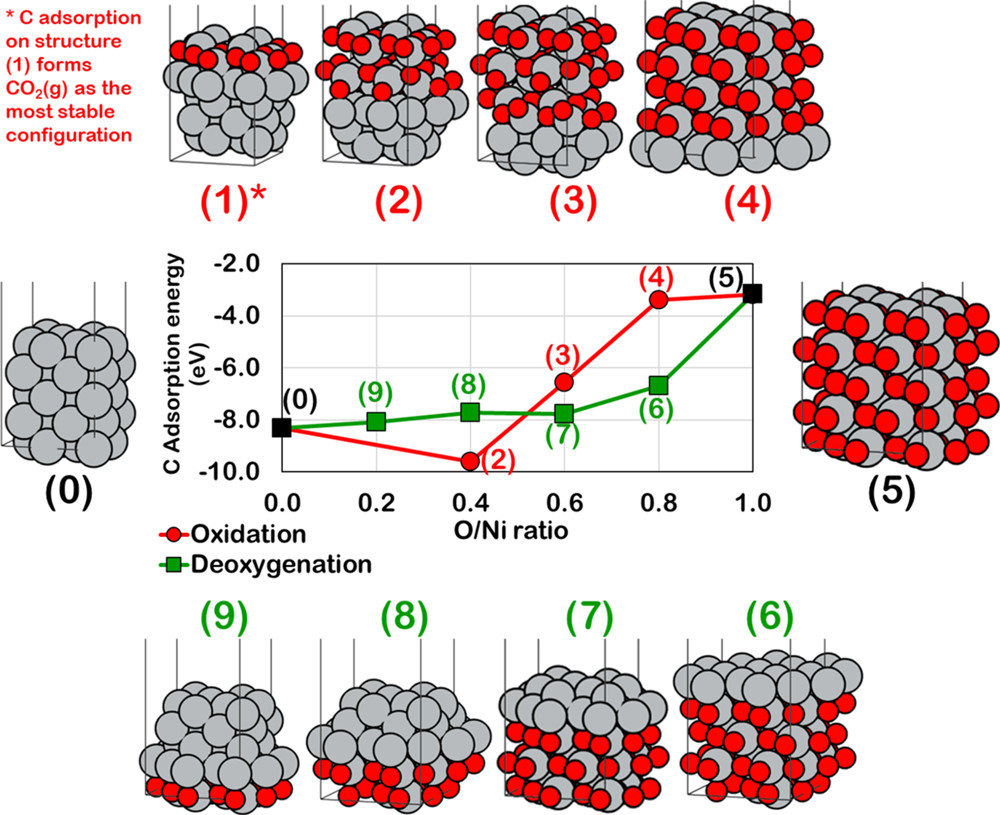
Supareak Praserthdam, Siriwimol Somdee, Meena Rittiruam, and Perla B. Balbuena, “Computational Study of the Evolution of Ni-Based Catalysts during the Dry Reforming of Methane,” Energy & Fuels, 34, 4, 4855-4864, (2020).
We evaluated the Ni-based catalyst surface properties during possible transformation pathways between its metallic, oxide, and carbide phases, causing catalytic deactivation. The study uses density functional theory (DFT) calculations to determine thermodynamics and reaction mechanisms of elementary reactions, and the ratings concept is introduced previously as an evaluation tool for the dry reforming reaction of methane (DRR) catalyst. The results for carbon atom adsorption strength and activation energy of higher coke formation (2C* ⇄ C–C* + *) suggest that on metallic surfaces, coke formation would be easy on the (111) facet but suppressed on the (100). Likewise, the carbide surface exposing metal atoms strongly binds to carbon and easily forms higher coke. In contrast, the oxide of Ni exhibits coke-resistant properties as it weakly adsorbs carbon. Finally, a ternary contour plot featuring metallic/oxide/carbide phases of Ni on the (111) facet was employed for identifying surface compositions, yielding highly reactive and stable DRR catalysts through a microkinetics model. It is found that, to become coke-resistant, the surface should contain less than 10% of carbide, whereas more than 75% of metallic surface is needed for the catalyst to be out of the coke formation zone, and to enter the coke removal zone, up to 80% of the metallic surface is required.
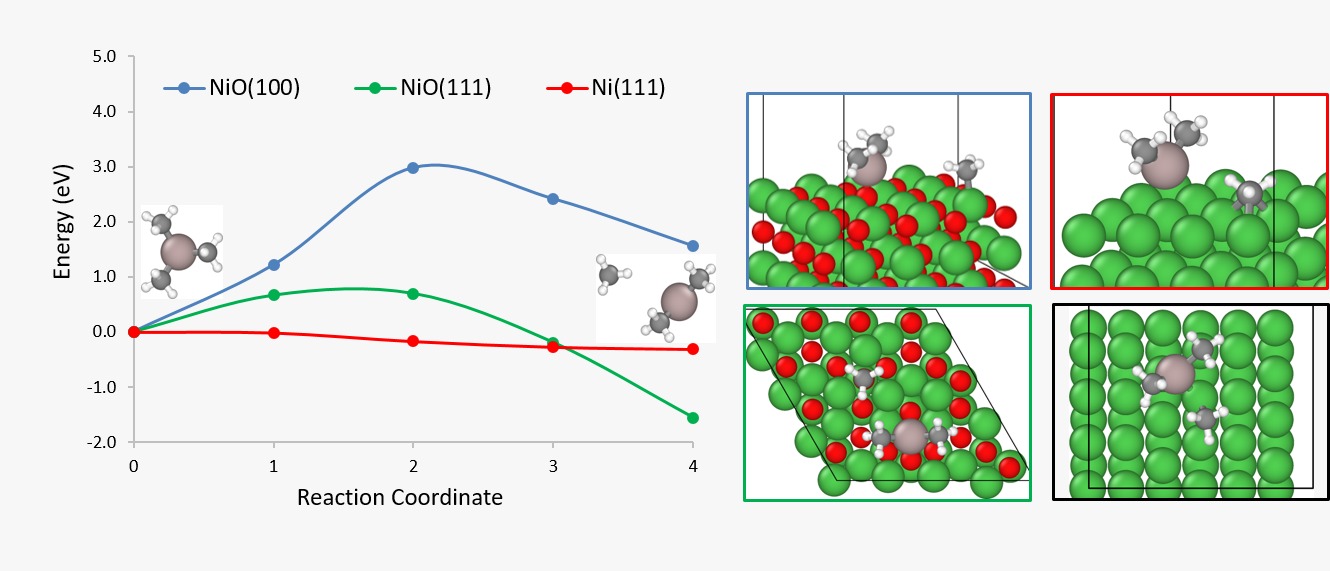
F. A. Ospina-Acevedo, S. Perez Beltran, P. B. Balbuena, “Mechanisms of Alumina Growth via Atomic Layer Deposition on Nickel Oxide and Metallic Nickel Surfaces,” Phys. Chem. Chem. Phys., 21, 24543-24553, (2019).
We aim at elucidating the mechanism of the trimethyl aluminum (TMA) decomposition on oxidized nickel (NiO) and metallic nickel (Ni) facets in the absence of a source of hydroxyl groups. This TMA decomposition mechanism constitutes the earliest stage of growth of Al2O3 coatings with the atomic layer decomposition (ALD) method, which stabilizes nickel catalysts in energy-intensive processes such as the dry reforming of methane. Our first-principles calculations suggest thermodynamic favorability for the TMA decomposition on metallic nickel compared to oxidized nickel. Moreover, the decomposition of TMA on metallic nickel showed almost no differences in terms of energy barriers between flat and stepped surfaces. Regarding the impact of the CH3 radicals formed after TMA decomposition, we calculated stronger adsorption on metallic nickel facets than on oxidized nickel, and these adsorption energies are comparable to the adsorption energies calculated in earlier works on Al2O3 ALD growth on palladium surfaces. These results lead us to believe in the growth of porous Al2O3 coatings triggered by CH3 contamination rather than due to preferential TMA decomposition on stepped and/or defective facets. The CH3 radicals are likely to be thermally stable at temperatures used during Al2O3 ALD processes, partially passivating the surface towards further TMA decomposition.
