
SELECT A TOPIC:
Beyond Li-ion battery technologies
Cell performance
Metal oxide cathodes
Lithium-Sulfur Batteries
SPAN
Sodium-Sulfur batteries
Electrocatalysis
Neuromorphic materials
2-dimensional materials
SWCNT
Beyond Li-ion battery technologies
Solid-electrolyte interphase (SEI) in Li metal anodes
click images to enlarge:
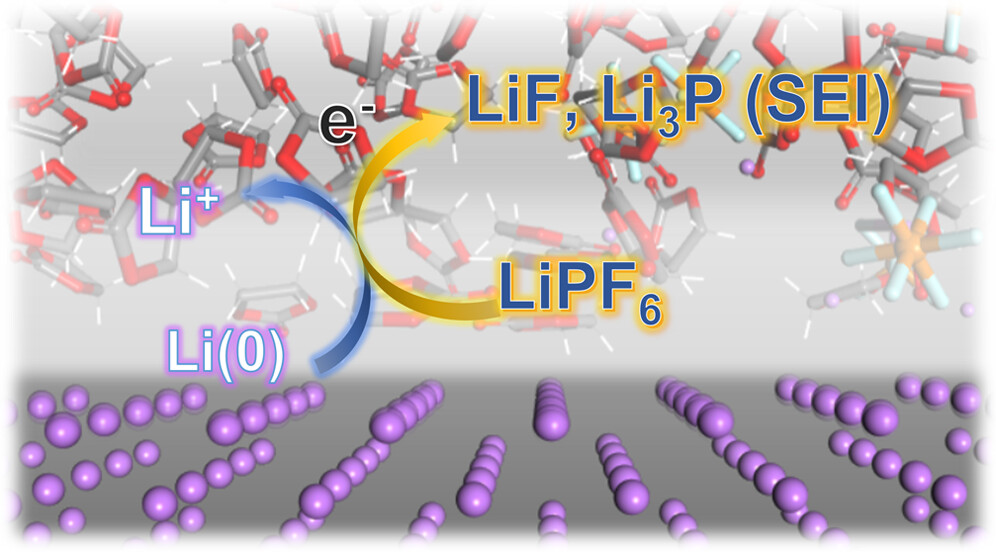
Dacheng Kuai and Perla B. Balbuena, “Inorganic Solid Electrolyte Interphase Engineering Rationales Inspired by Hexafluorophosphate Decomposition Mechanisms,” J. Phys. Chem. C, 127, 1744-1751, (2023).
Solid electrolyte interphase (SEI) engineering is an efficient approach to enhancing the cycling performance of lithium metal batteries. Lithium hexafluorophosphate (LiPF6) is a popular electrolyte salt. Mechanistic insights into its degradation pathways near the lithium metal anode are critical in modifying the battery electrolyte and SEI. In this work, we elucidate plausible reaction pathways in multiple representative electrolyte systems. Through ab initio molecular dynamics simulations, lithiation and electron transfer are identified as the triggering factors for LiPF6 degradation. Meanwhile, we find that lithium morphology and charge distribution substantially impact the interfacial dissociation pathways. Thermodynamic evaluation of the solvation effects shows that higher electrolyte dielectric constant and lithiation extent profoundly assist the LiPF6 decomposition. These findings offer quantitative thermodynamic and electronic structure information, which promotes rational SEI engineering and electrolyte tuning for lithium metal anode performance enhancement.
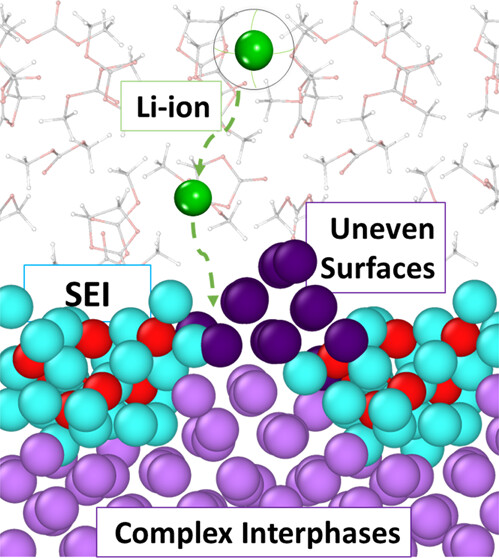
Stefany Angarita-Gomez and Perla B. Balbuena, “Lithium-ion Transport through Complex Interphases in Lithium Metal Batteries,” ACS Appl. Mater. Interfaces,14, 52, 56758-56766, (2022).
Lithium metal is one of the best anode candidates for next-generation batteries. However, there are still many unknowns regarding the structure and properties of the solid electrolyte interphase (SEI) formed due to electron transfer reactions between the Li metal surface and the electrolyte. In addition, because of the difficulties to study amorphous and dynamic phases and interphases, there are many questions about the ion diffusion mechanism through complex multicomponent materials and interphases. In this study, using first-principles theory and computation, we focus on developing a better understanding of the ion motion mechanisms in the vicinity of a SEI formed when a seed Li2O or LiOH cluster nucleates on the Li metal surface. We study the role of charge transfer at the interface between charged surfaces and the electrolyte, and we investigate the evolution of inhomogeneous Li metal deposits present in the neighborhood of the SEI nuclei, aiming to fundamentally understand how these events modify the ion transport through complex electrochemically active materials.
Saul Perez Beltran and Perla B. Balbuena, “SEI Formation Mechanisms and Li+ Dissolution in Lithium Metal Anodes: Impact of the Electrolyte Composition and the Electrolyte-to-Anode Ratio,” J. Power Sources, 551, 232203, (2022).
The lithium metal battery is one of today’s most promising high-energy-density storage devices. Its full-scale implementation depends on solving operational and safety issues intrinsic to the Li metal high reactivity leading to uncontrolled electrolyte decomposition and uneven Li deposition. In this work, we study the spontaneous formation of the solid electrolyte interphase (SEI) upon contact of Li metal with the electrolyte and describe the heterogeneous SEI morphological features. Multiple electrolyte formulations based on lithium bis(fluorosulfonyl)imide (LiFSI), dimethoxyethane (DME), dimethyl carbonate (DMC), 1,2,2-tetrafluoroethyl-2,2,3,3-tetrafluoropropyl ether (TTE) and bis(2,2,2-trifluoroethyl) ether (BTFE) are used. Findings include the description of the SEI evolution from dispersed LiO, LiS, LiN, and LiF clusters to a continuous and compact inorganic phase in which the LiO and LiF content depend on the presence of fluorine diluents. The role of the DME ether solvent helping the growth of a “wet-SEI” is compared to that of the highly unstable carbonate DMC, which decomposes into complex radical oligomers that might contribute to further electrolyte decomposition. The impact of the electrolyte-to-anode ratio on LiFSI decomposition is highlighted. Finally, we suggest the existence of a critical LiFSI concentration and electrolyte-to-anode ratio that could potentially balance the rate of electrolyte depletion and lithium consumption.
Francisco Ospina-Acevedo, Ningxuan Guo, and Perla B. Balbuena, “Lithium Oxidation and Electrolyte Decomposition at Li-Metal/Liquid Electrolyte Interfaces,” J. Mater. Chem. A, 8, 17036-17055, (2020).
We examine the evolution of events occurring when a Li metal surface is in contact with a 2 M solution of a Li salt in a solvent or mixture of solvents, via classical molecular dynamics simulations with a reactive force field allowing bond breaking and bond forming. The main events include Li oxidation and electrolyte reduction along with expansion of the Li surface layers forming a porous phase that is the basis for the formation of the solid-electrolyte interphase (SEI) components. Nucleation of the main SEI components (LiF, Li oxides, and some organics) is characterized. The analysis clearly reveals the details of these physical–chemical events as a function of time, during 20 nanoseconds. The effects of the chemistry of the electrolyte on Li oxidation and dissolution in the liquid electrolyte, and SEI nucleation and structure are identified by testing two salts: LiPF6 and LiCF3SO3, and various solvents including ethers and carbonates and mixtures of them. The kinetics and thermodynamics of Li6F, the core nuclei in the LiF crystal, are studied by analysis of the MD trajectories, and via density functional theory calculations respectively. The SEI formed in this computational experiment is the “native” film that would form upon contact of the Li foil with the liquid electrolyte. As such, this work is the first in a series of computational experiments that will help elucidate the intricate interphase layer formed during battery cycling using metal anodes.
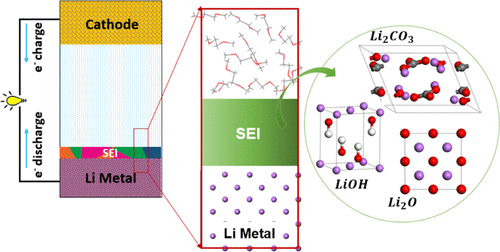
Ethan P. Kamphaus, Stefany Angarita, Xueping Qin, Minhua Shao, Mark Engelhard, Karl T. Mueller, Vijayakumar Murugesan, and Perla B. Balbuena, “Role of inorganic surface layer on solid electrolyte interphase evolution at Li-metal anodes,” ACS Appl. Mater. Interfaces, 11, 31467-31476, (2019).
Lithium metal is an ideal anode for rechargeable lithium-battery technology. However, the extreme reactivity of Li metal with electrolytes leads to solid electrolyte interphase (SEI) layers that often impede Li+ transport across interfaces. The challenge is to predict the chemical, structural, and topographical heterogeneities of SEI layers arising from a multitude of interfacial constituents. Traditionally, the pathways and products of electrolyte decomposition processes were analyzed with the basic and simplifying presumption of an initial pristine Li-metal surface. However, ubiquitous inorganic passivation layers on Li metal can reduce electronic charge transfer to the electrolyte and significantly alter the SEI layer evolution. In this study, we analyzed the effect of nanometric Li2O, LiOH, and Li2CO3 as surface passivation layers on the interfacial reactivity of Li metal, using ab initio molecular dynamics (AIMD) calculations and X-ray photoelectron spectroscopy (XPS) measurements. These nanometric layers impede the electronic charge transfer to the electrolyte and thereby provide some degree of passivation (compared to pristine lithium metal) by altering the redox-based decomposition process. The Li2O, LiOH, and Li2CO3 layers admit varying levels of electron transfer from a Li-metal slab and subsequent storage of the electronic charges within their structures. As a result, their ability to transfer electrons to the electrolyte molecules, as well as the extent of decomposition of bis(trifluoromethanesulfonyl)imide anions, is significantly reduced compared to similar processes on pristine Li metal. The XPS experiments revealed that when Li2O is the major component on the altered surface, LiF phases formed to a greater extent. The presence of a dominant LiOH layer, however, results in enhanced sulfur decomposition processes. From AIMD studies, these observations can be explained based on the calculated quantities of electronic charge transfer found for each of the passivating films.
SEI properties
click images to enlarge:
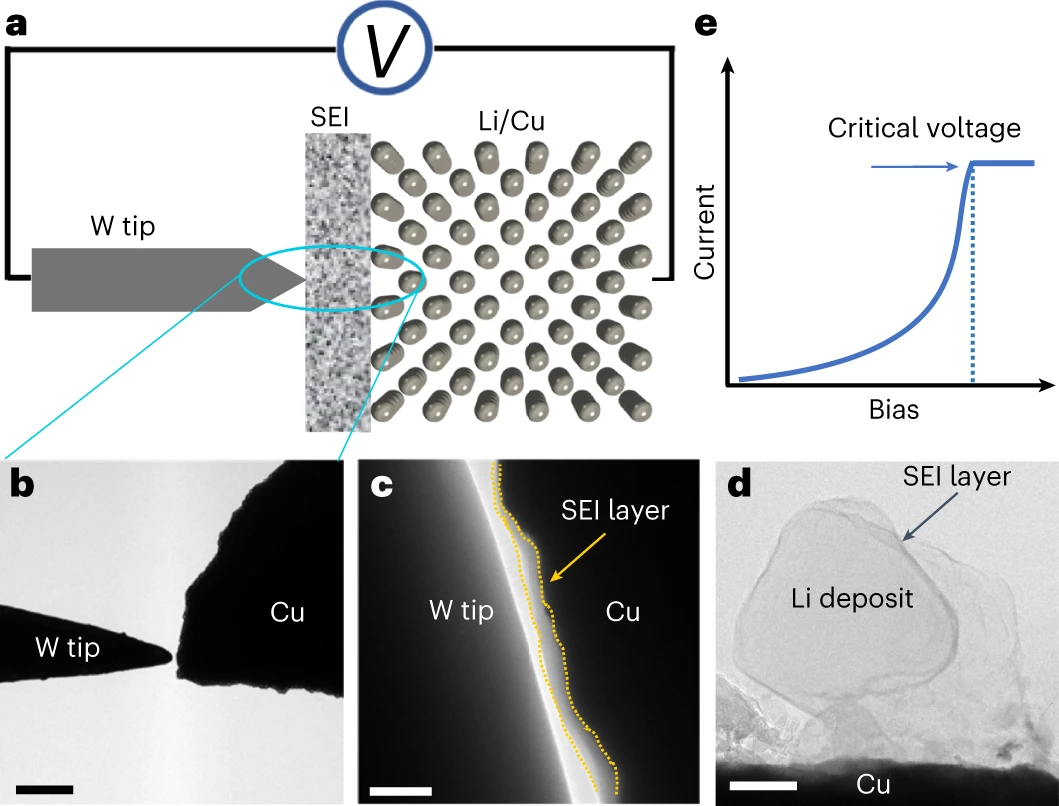
Yaobin Xu, Hao Jia, Peiyuan Gao, Diego E. Galvez-Aranda, Saul Perez Beltran, Xia Cao, Phung M. L. Le, Jianfang Liu, Mark H Engelhard, Shuang Li, Gang Ren, Jorge M. Seminario, Perla B. Balbuena, Ji-Guang Zhang, Wu Xu, Chongmin Wang, “Direct in-situ measurement of electrical properties of solid electrolyte interphase on lithium metal anode” Nature Energy, 8, 1345–1354 (2023)
The solid–electrolyte interphase (SEI) critically governs the performance of rechargeable batteries. An ideal SEI is expected to be electrically insulative to prevent persistently parasitic reactions between the electrode and the electrolyte and ionically conductive to facilitate Faradaic reactions of the electrode. However, the true nature of the electrical properties of the SEI remains hitherto unclear due to the lack of a direct characterization method. Here we use in situ bias transmission electron microscopy to directly measure the electrical properties of SEIs formed on copper and lithium substrates. We reveal that SEIs show a voltage-dependent differential conductance. A higher rate of differential conductance induces a thicker SEI with an intricate topographic feature, leading to an inferior Coulombic efficiency and cycling stability in Li||Cu and Li||LiNi0.8Mn0.1Co0.1O2 cells. Our work provides insight into the targeted design of the SEI with desired characteristics towards better battery performance.
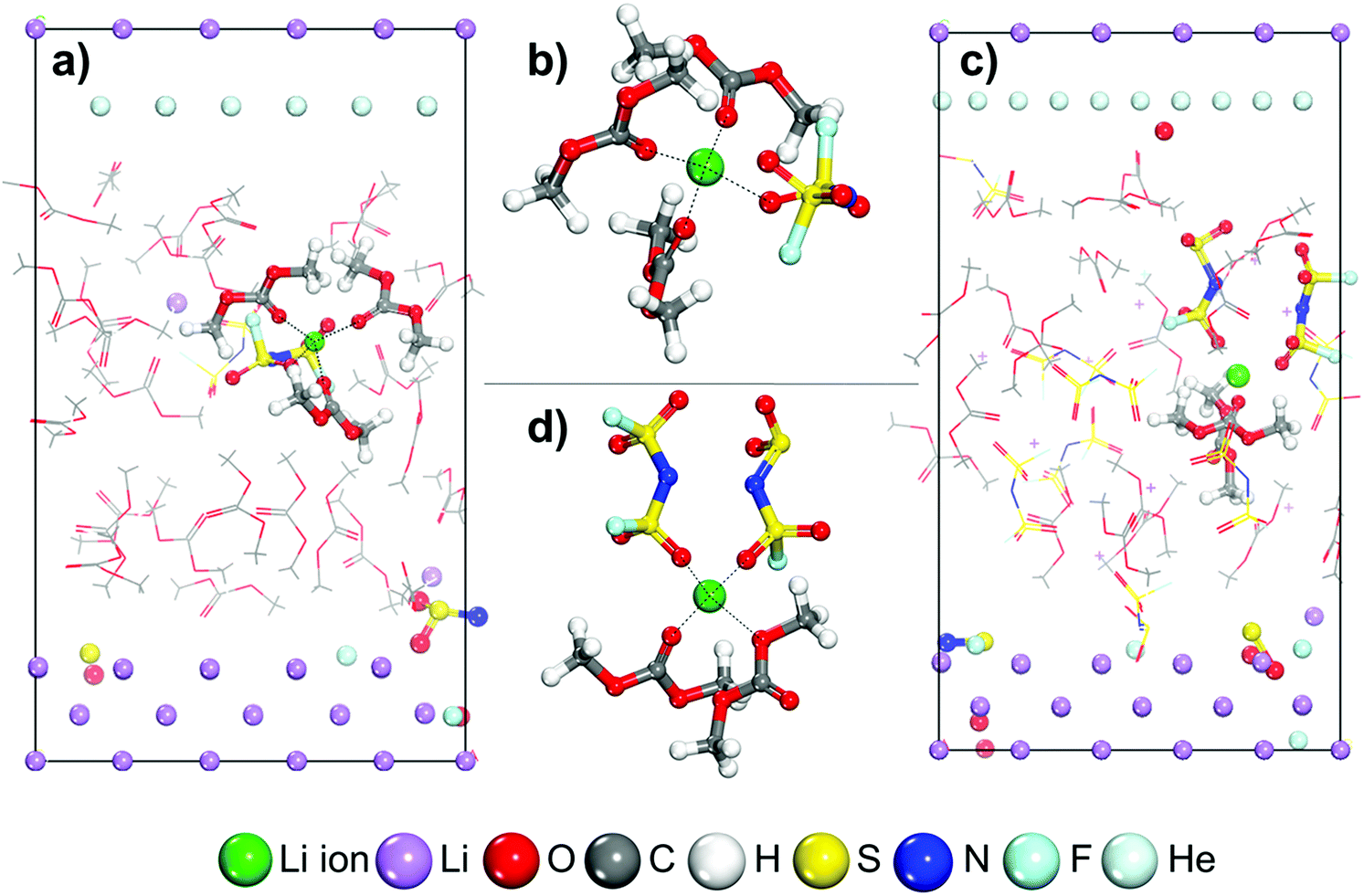
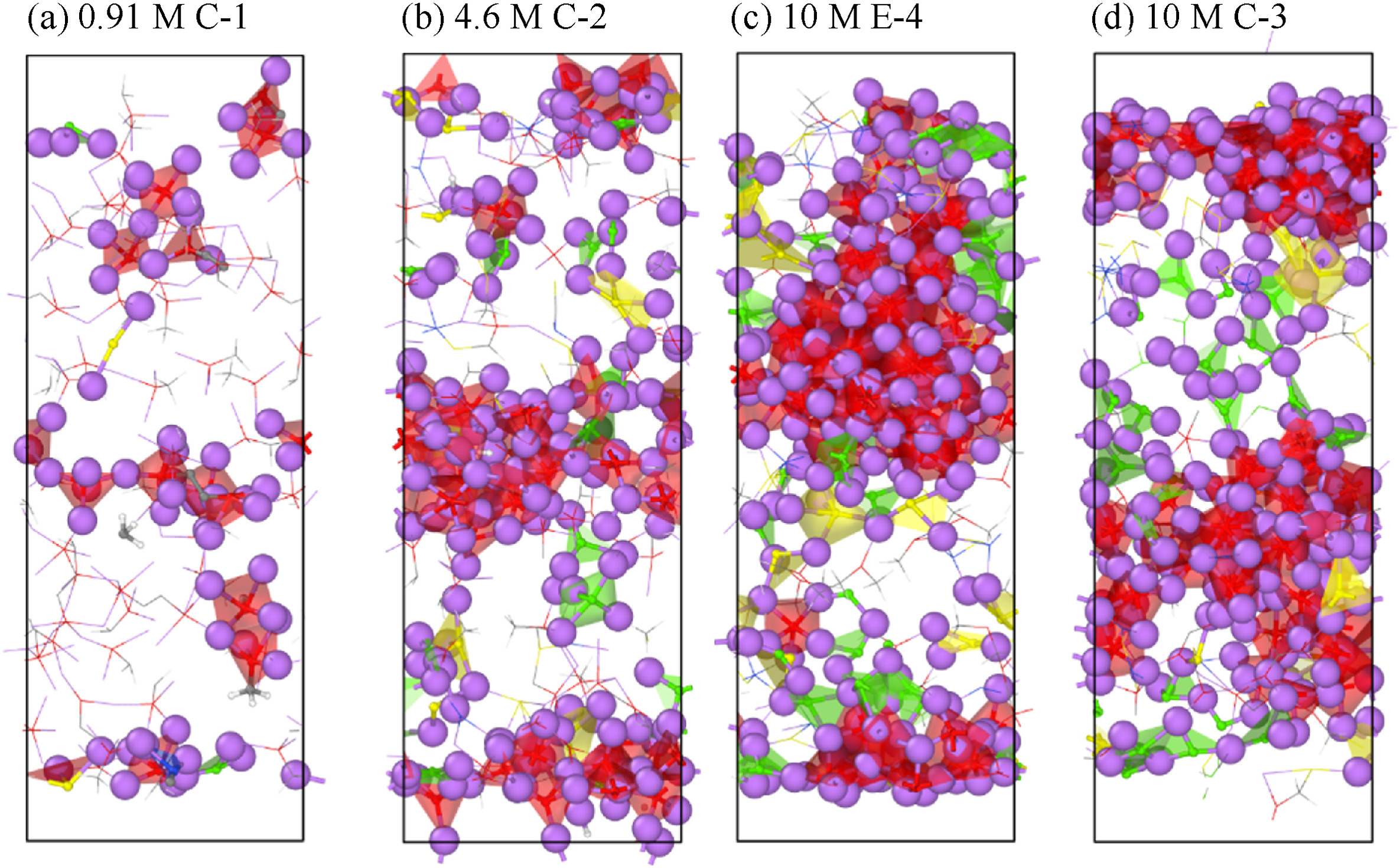
Stefany Angarita-Gomez and Perla B. Balbuena, “Ion Mobility and Solvation Complexes at Liquid-Solid Interfaces in Dilute, High Concentrated, and Localized High Concentrated Electrolytes,” Mater. Adv., 3, 6352-6363, (2022).
The underlying mechanisms of the solvated lithium cation diffusion and deposition on the Li metal surface occurring at electrochemical interfaces are still not fully understood. In this work, density functional theory and a thermodynamic integration method implemented in constrained-ab initio molecular dynamics are used to calculate the free energy profile for the lithium cation transport pathway in the absence of an external field. The trajectory and evolution of the solvation complex surrounding the lithium cation, alongside the effect of salt concentration and diluent presence are studied in carbonate-based electrolytes including low concentration electrolytes (LCEs), high concentration electrolytes (HCEs), and localized high concentration electrolytes (LHCEs). Energy barriers for transport and desolvation are obtained with the thermodynamic integration method and discussed in relation to the solvation shell surrounding the Li-ion. In dilute electrolytes, the energy barriers for cation diffusion in the electrolyte phase are relatively low and the final deposition is guided mostly by solvent reduction. In HCEs, the high connectivity between the primary solvation complex and the rest of the electrolyte leads to a significant increase in the energy barriers for diffusion, and the ion can get trapped in the electrolyte slowing down the deposition, while the early development of SEI formation shows a thick and compact SEI structure built by anion decomposition. In the LHCE the diluent helps in reducing the barriers found in HCEs and breaking the high connectivity thus facilitating cation diffusion and the simultaneous SEI formation process.
SEI evolution during cycling
click images to enlarge:
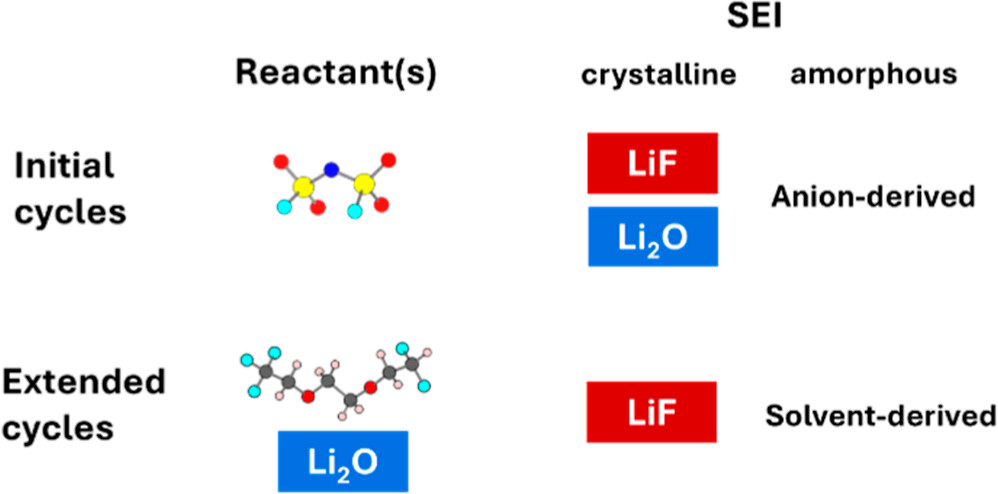
Sha Tan, Dacheng Kuai,, Zhiao Yu, Saul Perez-Beltran, Muhammad Mominur Rahman, Kangxuan Xia, Nan Wang, Yuelang Chen, Xiao-Qing Yang, Jie Xiao, Jun Liu, Yi Cui, Zhenan Bao, Perla B. Balbuena, Enyuan Hu, “Evolution and Interplay of Lithium Metal Interphase Components Revealed by Experimental and Theoretical Studies,” J. Am. Chem. Soc. 2024, 146, 17, 11711–11718,
Lithium metal batteries (LMB) have high energy densities and are crucial for clean energy solutions. The characterization of the lithium metal interphase is fundamentally and practically important but technically challenging. Taking advantage of synchrotron X-ray, which has the unique capability of analyzing crystalline/amorphous phases quantitatively with statistical significance, we study the composition and dynamics of the LMB interphase for a newly developed important LMB electrolyte that is based on fluorinated ether. Pair distribution function analysis revealed the sequential roles of the anion and solvent in interphase formation during cycling. The relative ratio between Li2O and LiF first increases and then decreases during cycling, suggesting suppressed Li2O formation in both initial and long extended cycles. Theoretical studies revealed that in initial cycles, this is due to the energy barriers in many-electron transfer. In long extended cycles, the anion decomposition product Li2O encourages solvent decomposition by facilitating solvent adsorption on Li2O which is followed by concurrent depletion of both. This work highlights the important role of Li2O in transitioning from an anion-derived interphase to a solvent-derived one.
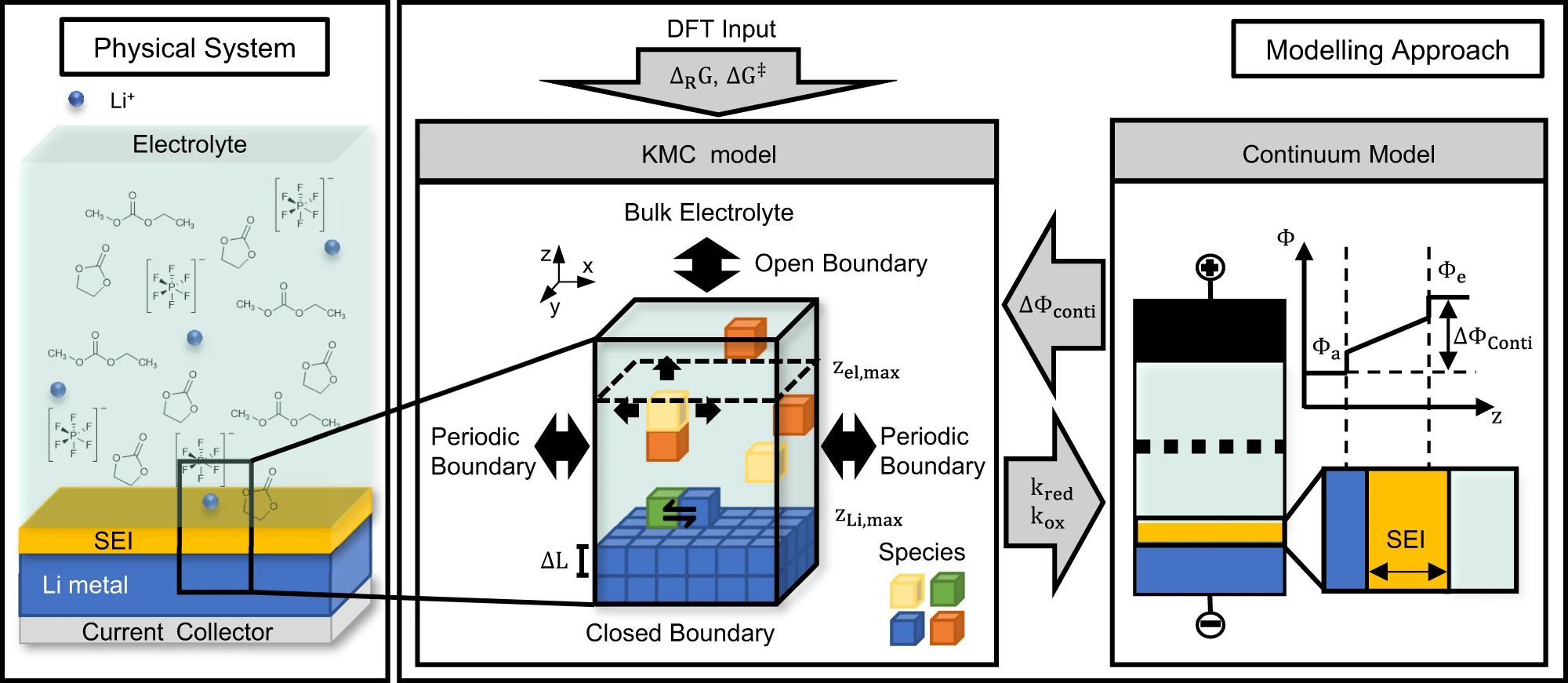
Janika Wagner, Dacheng Kuai, Michail Gerasimov, Fridolin Röder, Perla B. Balbuena, Ulrike Krewer, “Knowledge-driven design of Solid-Electrolyte Interphases on lithium metal: Insights from multiscale modelling,” Nat. Comm., 14, 1, 6823, (2023).
Due to its high energy density, lithium metal is a promising electrode for future energy storage. However, its practical capacity, cyclability and safety heavily depend on controlling its reactivity in contact with liquid electrolytes, which leads to the formation of a solid electrolyte interphase (SEI). In particular, there is a lack of fundamental mechanistic understanding of how the electrolyte composition impacts the SEI formation and its governing processes. Here, we present an in-depth model-based analysis of the initial SEI formation on lithium metal in a carbonate-based electrolyte. Thereby we reach for significantly larger length and time scales than comparable molecular dynamic studies. Our multiscale kinetic Monte Carlo/continuum model shows a layered, mostly inorganic SEI consisting of LiF on top of Li2CO3 and Li after 1 µs. Its formation is traced back to a complex interplay of various electrolyte and salt decomposition processes. We further reveal that low local Li+ concentrations result in a more mosaic-like, partly organic SEI and that a faster passivation of the lithium metal surface can be achieved by increasing the salt concentration. Based on this we suggest design strategies for SEI on lithium metal and make an important step towards knowledge-driven SEI engineering.
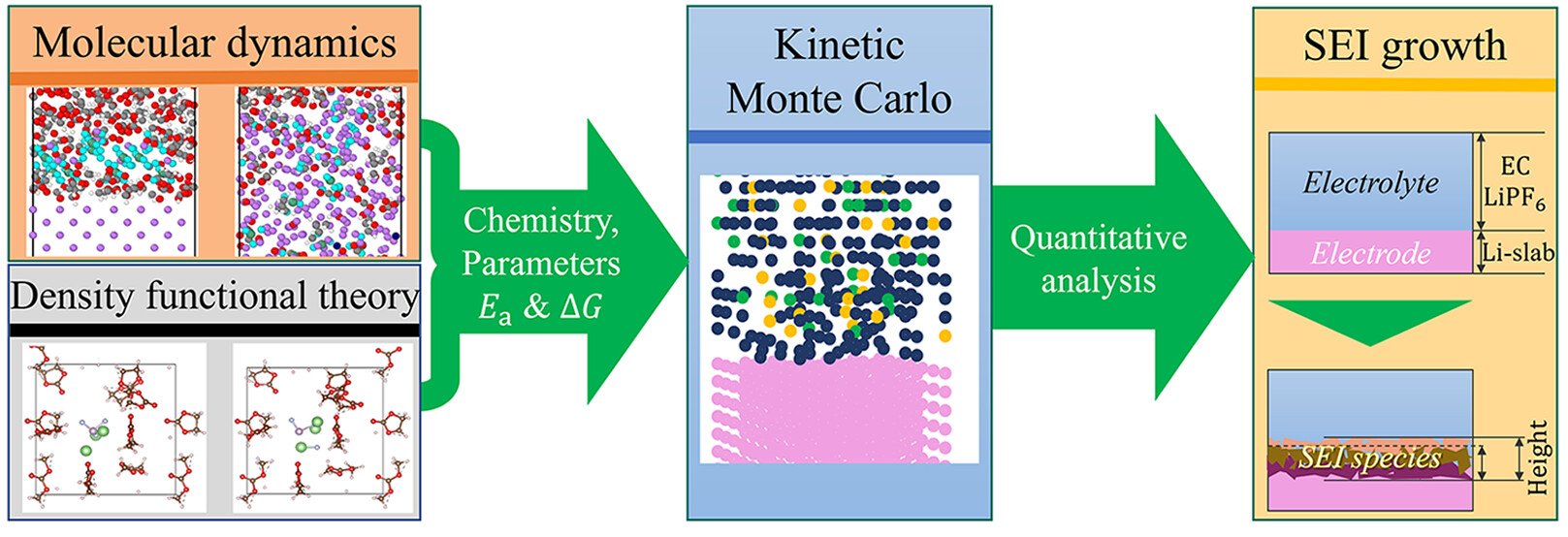
Michail Gerasimov, Fernando A. Soto, Janika Wagner, Florian Baakes, Ningxuan Guo, Francisco Ospina-Acevedo, Fridolin Röder, Perla B. Balbuena, Ulrike Krewer, “Species Distribution during Solid Electrolyte Interphase Formation on Lithium Using MD/DFT-Parameterized Kinetic Monte Carlo Simulations,” J. Phys. Chem. C, 127, 4872=4886, (2023).
Lithium metal batteries are one of the promising technologies for future energy storage. One open challenge is the generation of a stable and well performing Solid Electrolyte Interphase (SEI) between lithium metal and electrolyte. Understanding the complex interaction of reactions at the lithium surface and the resulting SEI is crucial for knowledge-driven improvement of the SEI. This study reveals the internal species distribution and geometrical aspects of the native SEI during formation by model-based analysis. To achieve this, a combination of molecular dynamics, density functional theory, and stand-alone 3D-kinetic Monte Carlo simulations is used. The kinetic Monte Carlo model determines the SEI growth features over a long time and length scale so that the SEI can be analyzed quantitatively. The simulation confirms the frequently postulated layered SEI structure arising from the decomposition of an ethylene carbonate/lithium hexafluorophosphate (2 M) electrolyte with lithium metal. These layers are not clearly separated, which is contrary to what is often reported. The gradient distribution of the species within the SEI therefore corresponds to a partly mosaic structured SEI at the borders of the layers. At the lithium surface, an inorganic layer of lithium fluoride and then lithium carbonate is observed, followed by an organic, more porous SEI layer consisting of lithium ethylene dicarbonate. Simulations further reveal the strong prevalence of corrosion processes of the metal, which provide more than 99% of the lithium for the SEI reaction processes. The salt contributes less than 1% to the SEI formation. Additionally, SEI formation below and above the initial interface was observable. The here presented novel modeling approach allows an unprecedented in-depth analysis of processes during native SEI formation that can be used to improve design for high battery performance and durability.
Electrolyte effects
click images to enlarge:
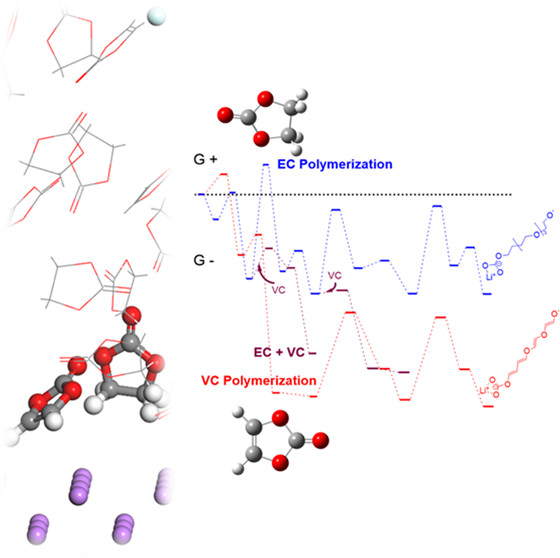
Dacheng Kuai and Perla B. Balbuena, “Solvent Degradation and Polymerization in the Li-Metal Battery: Organic Phase Formation in Solid-Electrolyte Interphases,” ACS Applied Materials & Interfaces, 14, 2817-2824, (2022).
The products of solvent polymerization and degradation are crucial components of the Li-metal battery solid-electrolyte interphase. However, in-depth mechanistic studies of these reactions are still scarce. Here, we model the polymerization of common lithium battery electrolyte solvents─ethylene carbonate (EC) and vinylene carbonate (VC)─near the anode surface. Being consistent with the molecular calculation, ab initio molecular dynamic (AIMD) simulations reveal fast solvent decompositions upon contact with the Li anode. Additionally, we assessed the thermochemical impacts of decarboxylation reactions as well as the lithium bonding with reaction intermediates. In both EC and VC polymerization pathways, lithium bonding demonstrated profound catalytic effects while different degrees of decarboxylation were observed. The VC polymerization pathways with and without ring-opening events were evaluated systematically, and the latter one which leads to poly(VC) formation was proven to dominate the oligomerization process. Both the decomposition and polymerization reactivities of VC are found to be higher than EC, while the cross-coupling between EC and VC has an even lower-energy barrier. These findings are in good agreement with experimental evidence and explanatory toward the enhanced performance of VC-added lithium-metal batteries.
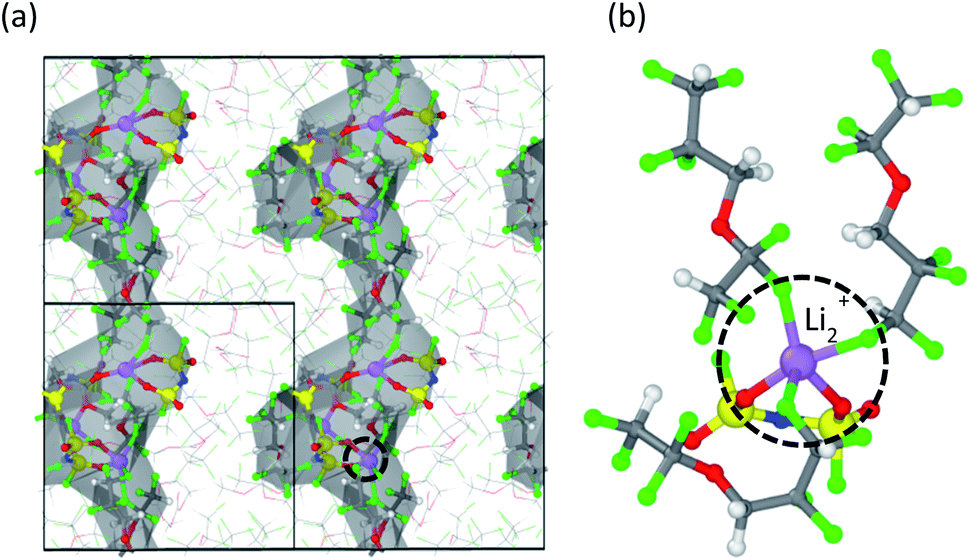
Saul Perez Beltran, Xia Cao, Ji-Guang Zhang, Patrick Z. El-Khoury, and Perla B. Balbuena, “Influence of Diluent Concentration in Localized High Concentration Electrolytes: Elucidation of Hidden Diluent-Li+ Interactions and Li+ Transport Mechanism,” J. Mater. Chem. A, 9, 17459-17473, (2021).
Localized high concentration electrolytes (LHCE) offer a viable dilution strategy for high concentration electrolytes (HCE) as the dilution process barely impacts the enhanced reductive/oxidative behavior of the HCE formulation but significantly lowers the overall viscosity and, in most cases, increases the ionic conductivity. On the other hand, experimental studies indicate that fluorinated ether electrolytes such as 1,1,2,2-tetrafluoroethylene 2,2,3,3-tetrafluoropropyl ether (TTE) help grow enhanced passivation layers on Ni-rich NMC cathodes. In this work, we study LHCE formulations based on lithium bis(fluorosulfonyl)imide (LiFSI), dimethyl carbonate (DMC), and TTE as the diluent. We use molecular dynamics methodologies, and Raman spectra measurements, to address to what extent the diluent content impacts the coordination behavior of the aggregated structures in the electrolyte and to evaluate the Li+ transport properties under the influence of an external electric field. In contrast to other fluorinated ethers, we find that TTE interacts with Li+via fluorine atoms, partially limiting the DMC–Li+ interactions hence altering the Li+ solvation coordination. This competitive interaction with Li+ between the organic solvent and the TTE diluent influences the electrolyte’s reductive/oxidative behavior. Nevertheless, the bonding strength of the Li+–FTTE is much weaker than those of the Li+–ODMC and Li+–OFSI−. Therefore, the existence of Li+–FTTE is in a transient state rather than in a steady state. These results provide plausible guiding rules for future dilution strategies of HCE electrolytes. We also demonstrate that Li+ ions drift under the electric field’s influence via repeated ion dissociation/association processes following a hopping conduction mechanism. Li+ ions jump between aggregated networks where Li–O interactions dominate via diluent-enriched phases, a process in which the solvation shells temporarily mutate to a Li–F dominated coordination structure. We expect our results to contribute an improved atomic-level understanding of the solvation structure and dynamics of LHCE electrolytes.
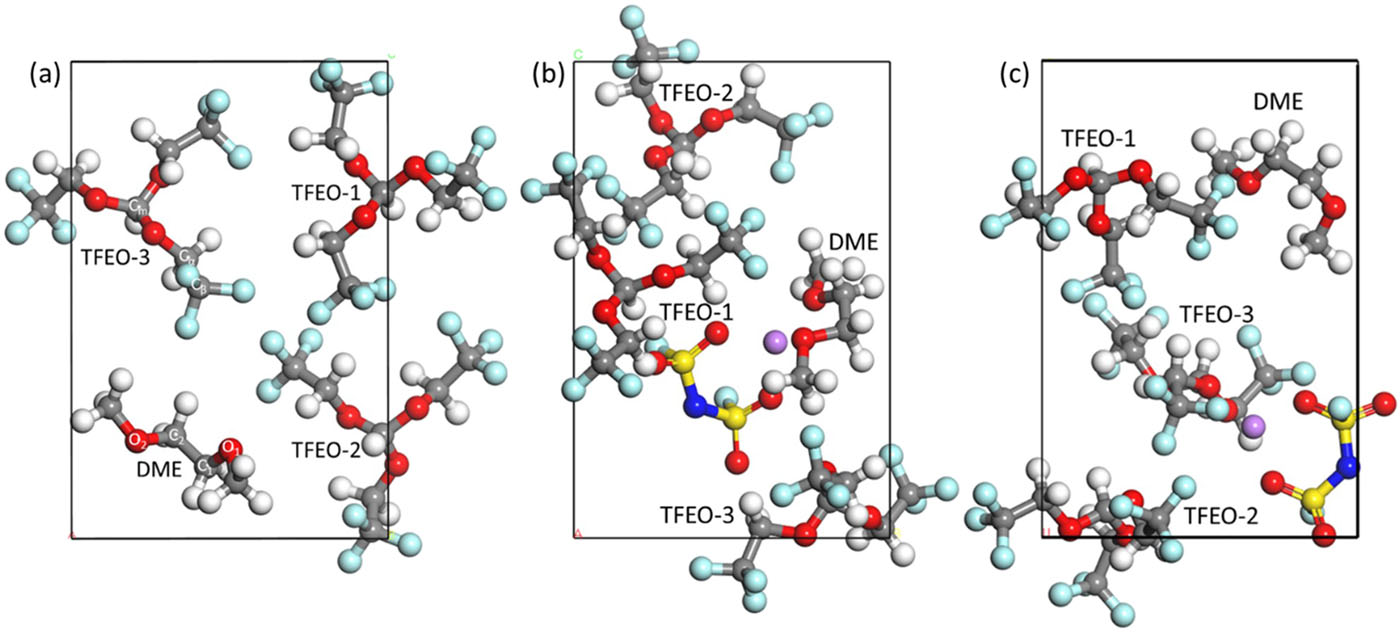
Yu Zheng and Perla B. Balbuena, “Localized High Concentration Electrolytes Decomposition under Electron-rich Environments“, J. Chem. Phys, 154, 104702 (2021).
Localized high concentration electrolytes have been proposed as an effective route to construct stable solid-electrolyte interphase (SEI) layers near Li-metal anodes. However, there is still a limited understanding of the decomposition mechanisms of electrolyte components during SEI formation. In this work, we investigate reactivities of lithium bis(fluorosulfonyl)imide (LiFSI, salt), 1,2-dimethoxyethane (DME, solvent), and tris(2,2,2-trifluoroethyl)orthoformate (TFEO, diluent) species in DME + TFEO mixed solvents and 1M LiFSI/DME/TFEO solutions. By supplying an excess of electrons into the simulation cell, LiFSI is initially reduced via a four-electron charge transfer reaction yielding F− and N(SO2)23−. The local solvation environment has little effect on the subsequent TFEO reaction, which typically requires 6 |e| to decompose into F−, HCOO−, CH2CF−, and −OCH2CF3. Besides, the TFEO dehydrogenation reaction mechanism under an attack of anions is also identified. Unlike salt and diluent, DME shows good stability with any excess of electrons. The energetics of most relevant reactions are characterized. Most reactions are thermodynamically favorable with low activation barriers.

Saul Perez Beltran, Xia Cao, Ji-Guang Zhang, and Perla B. Balbuena, “Localized High Concentration Electrolytes for High Voltage Lithium-Metal Batteries: Correlation between the Electrolyte Composition and its Reductive/Oxidative Stability,” Chem. Mater. 32, 5973-5984, (2020).
We demonstrate a first-principles screening methodology as an effective tool to explore electrolyte formulations for the new generation of high energy density rechargeable batteries. We study the liquid structure and electronic properties in dilute electrolytes, high concentration electrolytes (HCE), and localized high concentration electrolytes (LHCE), with focus on electrolyte formulations based on lithium bis(fluorosulfonyl)imide (LiFSI), dimethyl carbonate (DMC), and bis(2,2,2-trifluoroethyl) ether (BTFE) as a diluent. We describe the solvation complexes in the dilute electrolyte and explore structural changes triggered by the increase in lithium salt concentration for HCEs and the diluent effects in LHCEs. In HCE formulations, there is a 4-fold coordination environment of lithium-ions as in the dilute electrolyte, but the number of lithium-ion interactions with O atoms from FSI– anions dominates. In these solutions, the ability of the FSI– anions to interact with multiple lithium-ions allows complex 3D network formation and influences the reductive/oxidative behavior of the electrolyte. Interestingly, in LHCEs, the BTFE diluent molecules do not change the 3D solution structure when diluting the HCE formulation from 5.49 to 3.83 M. However, there is a composition threshold where the structural and electronic behavior may change. We show that diluting the HCE electrolyte with BTFE down to 1.77 M breaks the three-dimensional solution structure into an island-like solvation complex. We relate these structural changes to the electronic properties of the electrolytes finding a causal relationship between the reductive/oxidative behavior and the lithium–oxygen interaction mechanisms in the solvated complexes. The coordination with lithium-ions lowers the electrolyte LUMO and HOMO levels: the higher is the number of interactions with lithium-ions, the more likely the solvent molecule, FSI– anion, or diluent molecule is to be reduced and the less likely it is to become oxidized. The evolution of the solvated ion structure in HCE and LHCE suggests a close connection to a corresponding change in the lithium-ion transport mechanisms for these electrolytes.
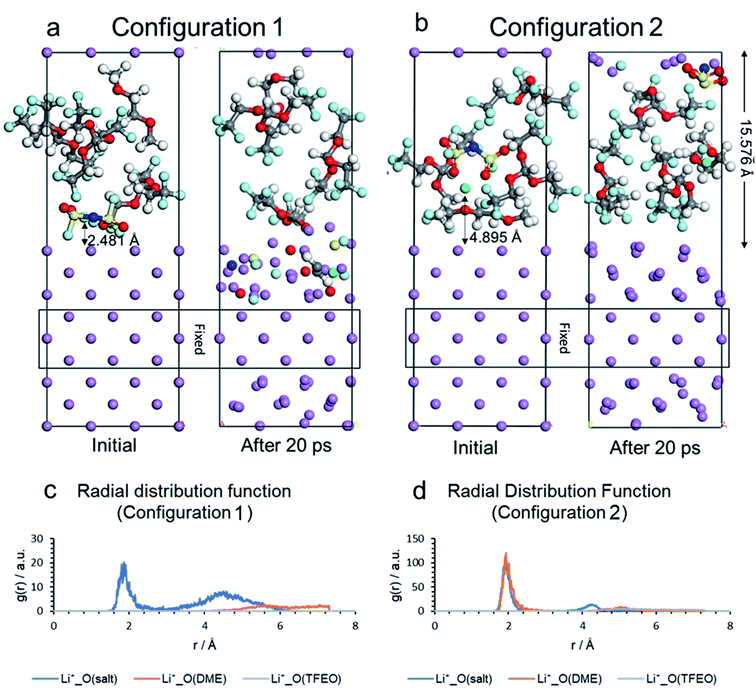
Yu Zheng, Fernando A. Soto, Victor Ponce, Jorge M. Seminario, Xia Cao, Ji-Guang Zhang, and Perla B. Balbuena, “Localized High Concentration Electrolyte Behavior near a Lithium-Metal Anode Surface,” J. Mater. Chem. A, 7, 25047-27055, (2019).
Wide-scale practical application of rechargeable lithium–metal batteries remains a significant challenge due to dendrite growth. To overcome this challenge, electrolytes must be designed to allow for the formation of protective solid electrolyte interphase (SEI) layers on the highly reactive lithium–metal anode (LMA) surfaces. Recently, novel localized high-concentration electrolytes (LHCEs) were introduced as a potential solution to enable dendrite-free cycling of LMAs, by using an inert solvent to “dilute” the high concentration electrolytes. Ideally, the diluent itself does not dissolve the salt but is miscible with the solvent to form a localized high concentrated salt/solvent cluster surrounded by the diluent. However, detailed structure and potential surface reactions that may take place in LHCE environment are not yet clear. In this work, we investigated the reactivity of 1 M lithium bis(fluorosulfonyl)imide (LiFSI) in a mixture of dimethoxyethane (DME)/tris(2,2,2-trifluoroethyl)orthoformate (TFEO) (1 : 3 by mol) electrolyte near a Li metal surface based on density functional theory and ab initio molecular dynamics (MD) simulations. Selected liquid interfacial configurations were obtained from classical MD simulations. Our results indicate that when salt and TFEO molecules are close to each other and to the surface, fluoride anions resulting from the fast salt anion decomposition can trigger a cascade of reactions that lead to the decomposition of TFEO. However, if the Li cation is initially solvated by DME and the anion forming a complex, the stability of the anion increases significantly. The Li solvated structure is implied in the LHCE concept; however statistically the larger amount of TFEO molecules suggest also the first scenario leading to TFEO decomposition. Therefore, the broader implication of our simulations is that the defluorination of TFEO may contribute, together with the anion decomposition, to the observed rapid formation of a stable SEI on the surface of the lithium metal; consequently, favorably affecting the stability of LMAs during battery operation.
Solvation effects at interfaces
click images to enlarge:
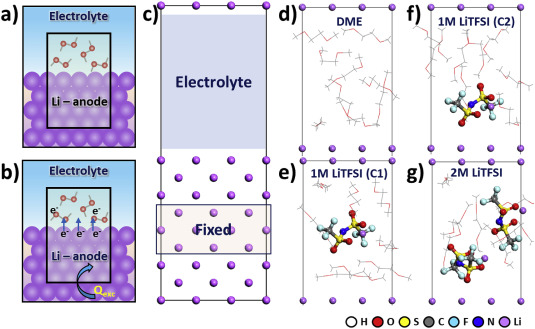
Stefany Angarita-Gomez and Perla B. Balbuena, “Solvation vs. Surface Charge Transfer: An Interfacial Chemistry Game Drives Cation Motion,” Chem. Comm., 57, 6189-6192, (2021)
Electrolyte structure and ion solvation dynamics determine ionic conductivities, and ion (de)solvation processes dominate interfacial chemistry and electrodeposition barriers. We elucidate electrolyte effects facilitating or impeding Li+ diffusion and deposition, and evaluate structural and energetic changes during the solvation complex pathway from the bulk to the anode surface.
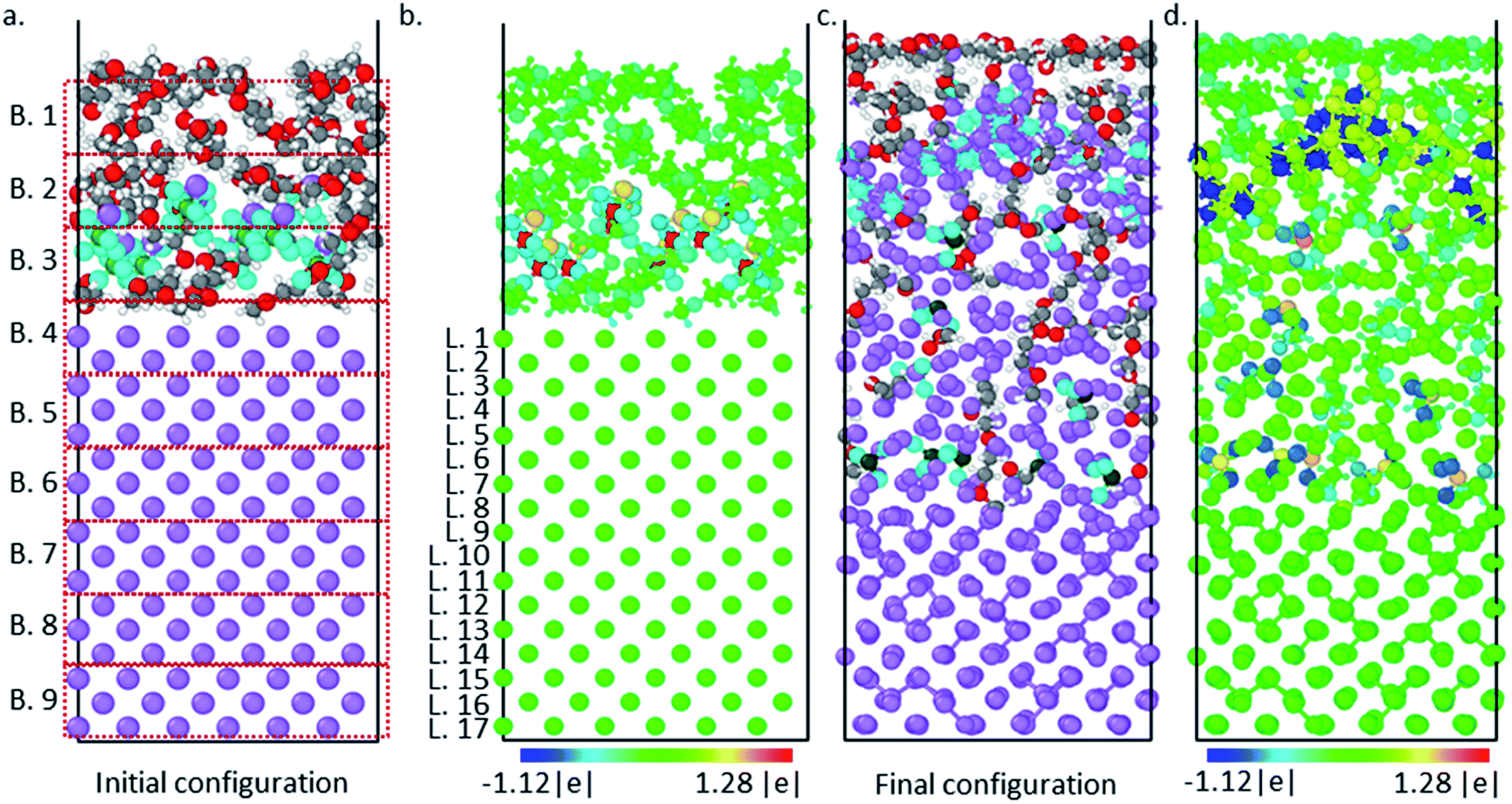
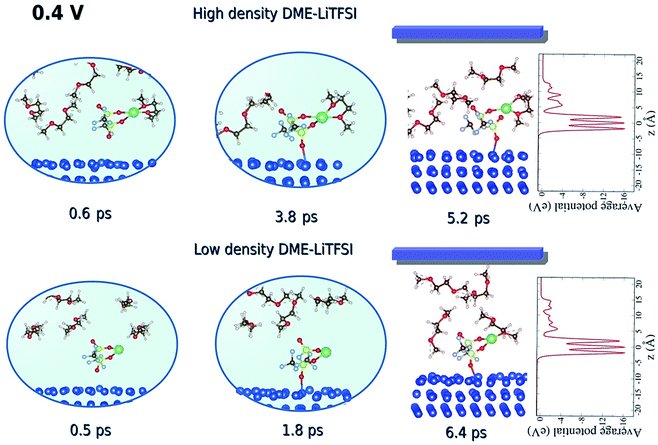
Luis E. Camacho-Forero and Perla B. Balbuena, “Effects of Charged Interfaces on Electrolyte Decomposition at the Lithium Metal Anode,” J. Power Sources, 472, 228449, (2020).
Lithium–Sulfur batteries are promising candidates to substitute conventional Li-ion batteries due to their higher energy density and reduced cost. However, several challenges related to the reactivity of the lithium metal anode have prevented this technology from becoming broadly commercialized. Lithium’s high reactive nature leads to the continuous decomposition of the electrolyte and the formation of the solid-electrolyte interphase (SEI) layer. Thus, a comprehensive understanding of how the SEI film is formed is crucial to help elucidate improvements for this battery technology. In this work, we use density functional theory (DFT) based computational methods to investigate the effect of charged interfaces on the electrolyte degradation at the lithium anode. Several electrolyte mixtures were considered including 1,2-dimethoxyethane (DME) and 1,3-dioxolane (DOL) as solvents, and lithium bis(fluorosulfonyl)imide (LiFSI) and lithium bis(trifluoromethanesulfonyl)imide (LiTFSI) as salts. It is found that the extent of salt decomposition is higher when the interface is charged. In addition, two types of solvent reduction mechanisms are identified: C–O bond cleavage and radical attacks. Charge transfer and evolution analysis are characterized in detail. Finally, solvent decomposition reactions are found to become more thermodynamically favorable and their activation barriers to be diminished under the effect of constant potentials and electric fields.
Roberto C. Longo, Luis E. Camacho-Forero, and Perla B. Balbuena, “Charge-Mediated Cation Deposition on Metallic Surfaces,” J. Mater. Chem. A, 7, 8527-8539, (2019).
This work reveals the general mechanisms of Li+ cation partial reduction and further deposition under specific electrolyte conditions, and in the proximity of an electrified metal surface. The factors affecting the ion complexation and transport and resultant adsorbed structures are identified for various external electric fields and for several applied voltages. Using ab initio methods, we investigate the relation between solvent–salt structures and dynamics, cation reductive stability, and the existing electric field between electrodes, or under the applied potential if the system is designed as an electrochemical cell. In absence of an applied field, it is found that not only cation but also non-solvated salt deposition on a metallic surface is an endothermic process. However, localized surface polarization orbitals created by an external electric field affect both the thermodynamics and kinetics of the adsorption, leading to a drastic change on the energetic profile; ab initio molecular dynamics simulations describe in detail the underlying mechanisms. Externally applied bias to an electrochemical cell also changes the deposition dynamics: by examining various solvent/salt combinations, we unravel the cation deposition mechanisms at both low and moderately high voltages, determining the relative time scale of each separate process. Different polyhedra formed by the metal cation and its surrounding oxygen atoms appear to be the key indicator driving deposition dynamics at different voltages. Our findings disclose the importance of selecting appropriate voltage windows for stable and uniform growth of metal anode layers, showing the mechanisms leading to impurity formation and non-uniform cation deposition, as well as the role of charged interfacial phenomena in designing stable electrode–electrolyte interfaces.
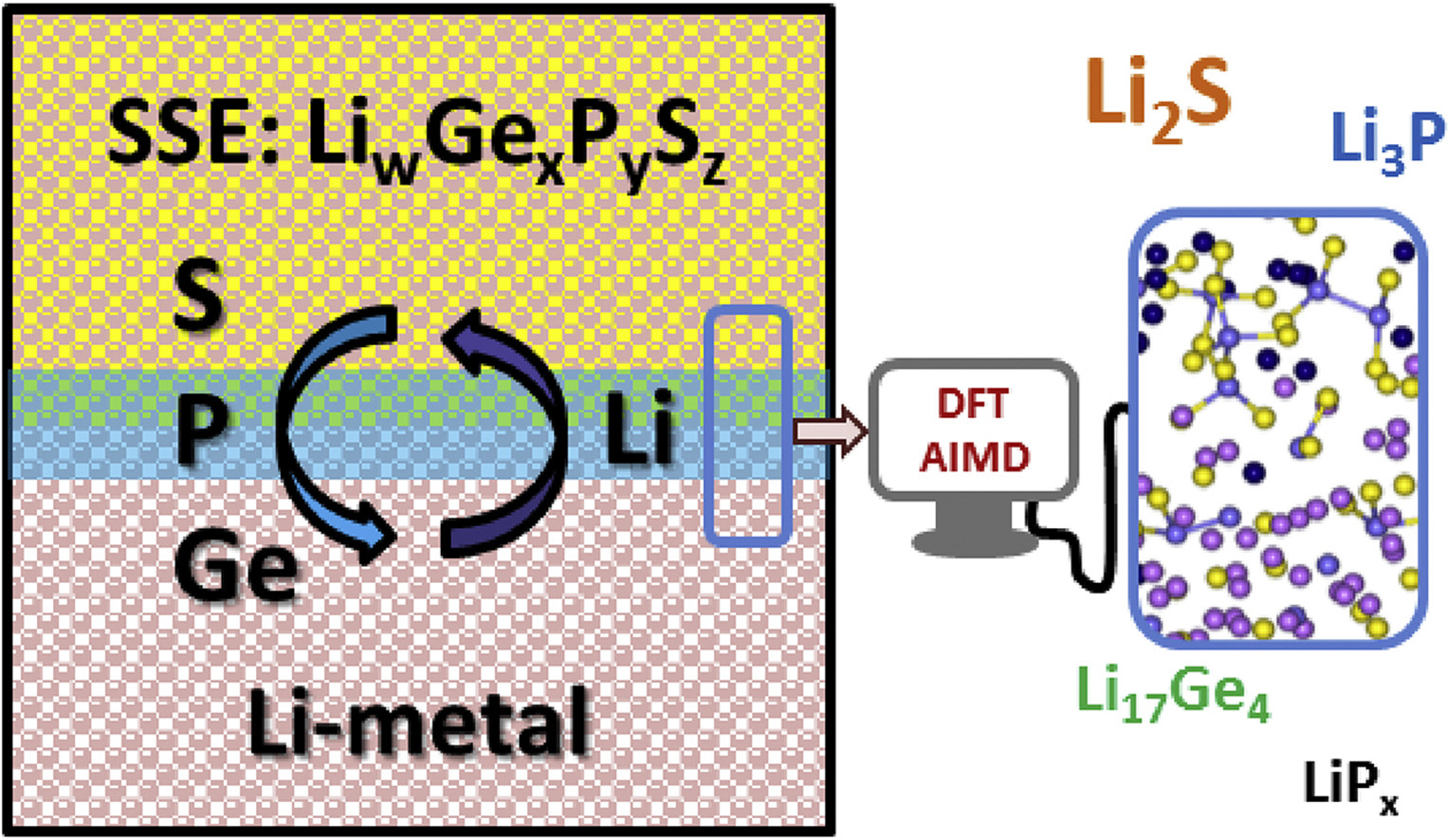
Luis E. Camacho-Forero and Perla B. Balbuena, “Exploring Interfacial Stability of Solid-State Electrolytes at the Lithium-Metal Anode Surface,” J. Power Sources, 396, 782-790, (2018).
Solid state electrolytes are promising materials to mitigate the issues derived from the extreme reactivity of the lithium metal anodes in Li-metal batteries. The main properties sought for this application are high ionic conductivity, low electronic conductivity, and high interfacial stability. Here we investigate a class of sulfides (Li10GeP2S12, Li2P2S6, β-Li3PS4, and Li7P3S11) that have shown relatively good ionic conductivities. However, little is known regarding their interfacial stability. We use density functional theory and ab initio molecular dynamics simulations to investigate the time evolution of the interfacial structure. We characterize atomic diffusion and reactions happening at the picosecond time scale, allowing us to identify the main interfacial products: Li2S, Li3P and Li17Ge4. We then study how the reactivity changes when the Li metal surface is coated with a thin film of Li2S.
