
2-dimensional materials
click images to enlarge:
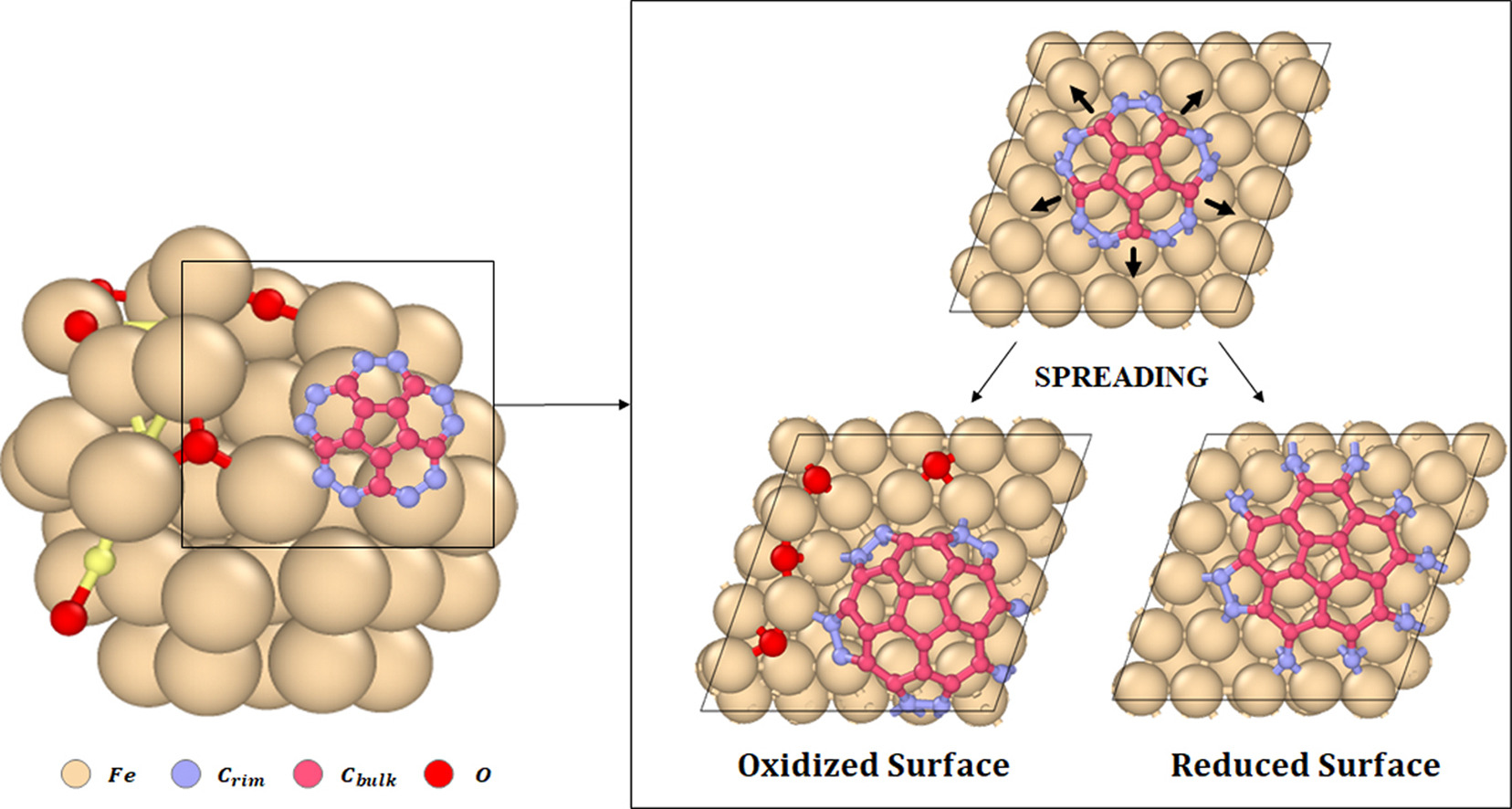
Mauricio C. Diaz and Perla B. Balbuena, “On the Role of Surface Oxygen during Nascent Single-Walled Carbon Nanotube Cap Spreading and Tube Nucleation on Iron Catalysts,” Carbon, 184, 470-478 (2021).
Single-walled carbon nanotubes (SWCNTs) are commonly grown in oxygen-rich environments. The precursor used as input for the SWCNT synthesis reaction typically provides oxygen atoms either as a pure gas or as an oxygen-containing compound. Similarly, metal oxide substrates are used to support the catalysts or as reactor wall materials and can transfer oxygen atoms while in contact with the catalytic particles. It has been proposed and experimentally observed that the interaction of the metal catalyst with specific promoters (e.g., oxygen, sulfur, hydrogen) triggers significant changes in the SWCNT properties during spreading, nucleation and growth. However, very few studies address the carbon-catalyst surface interactions in rich oxygen environments. Here we show that an increased surface oxygen concentration in the neighborhood of a spreading nascent carbon shell reduces the strength of interaction of the carbon shell with the iron catalyst’s surface and increases the probability of carbon nucleation and SWCNT lift-off. The evolution of the carbon-surface interaction during the spreading and nucleation phases suggests that closed pentagons at the rim are preferentially formed in oxidized iron surfaces, and they exhibit the strongest and most stable interaction with the iron surface.
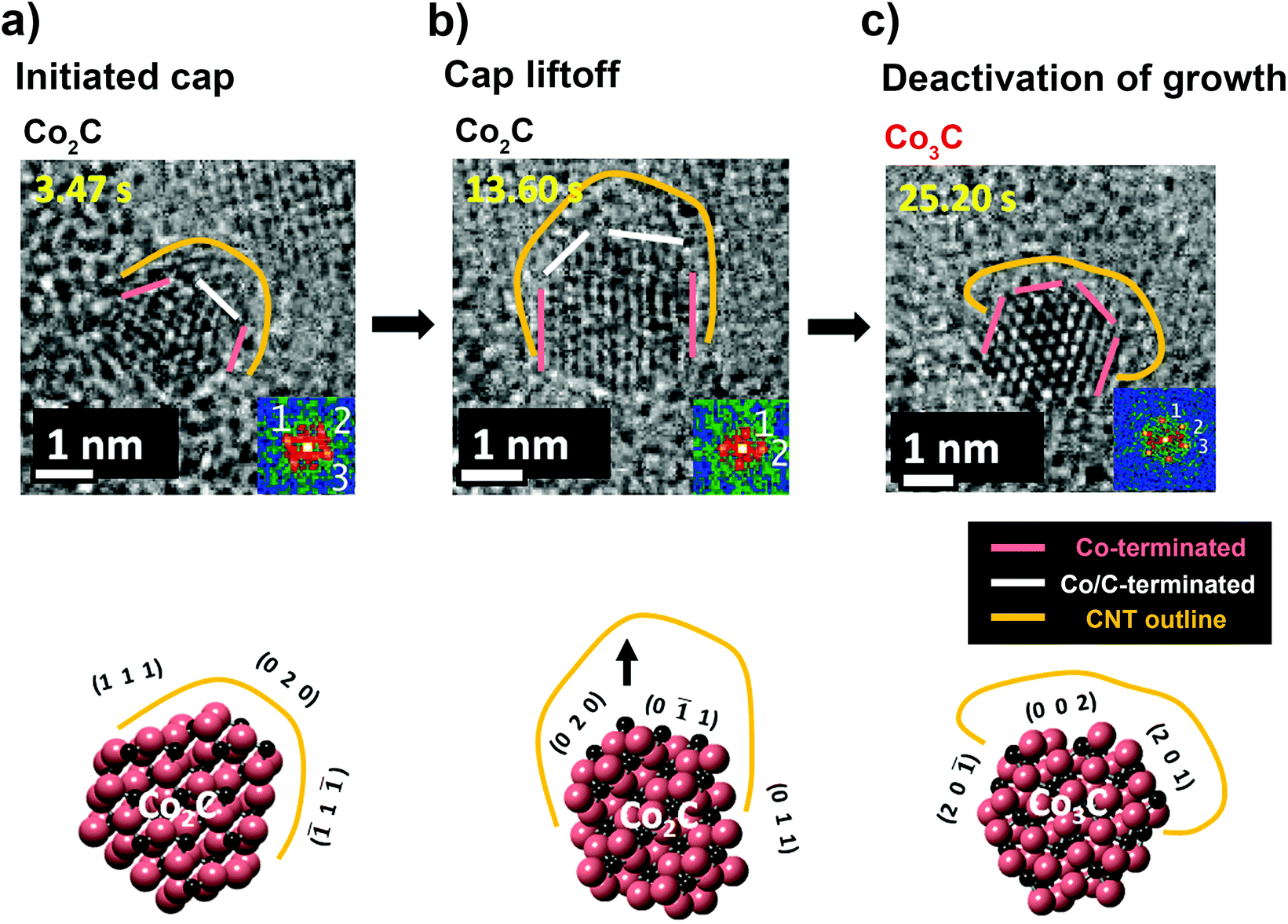
Hsin-Yun Chao, Hua Jiang, Francisco Ospina Acevedo, Perla B. Balbuena, Esko I. Kauppinen, John Cumings, and Renu Sharma, “Structure and Activity Relationship for Single-Walled Carbon Nanotube Growth Confirmed by In-Situ observations and Modeling,” Nanoscale, 12, 21923-21931 (2020).
The structure and phase transformation of a cobalt (Co) catalyst, during single walled carbon nanotube (SWCNT) growth, is elucidated for inactive, active and deactivated nanoparticles by in situ imaging using an environmental transmission electron microscope. During nanotube growth, the structure was analyzed using Miller indices to determine the types of planes that favor anchoring or liftoff of nanotubes from the Co catalyst. Density functional theory was further applied to model the catalyst interactions to compare the work of adhesion of the catalyst’s faceted planes to understand the interactions of different Miller planes with the graphene structure. Through in-depth studies of multiple distinct Co nanoparticles, we established a dominant nanoparticle phase for SWCNT growth. In addition, we identified the preferred lattice planes and a threshold for work of adhesion to allow the anchoring and liftoff of SWCNTs.
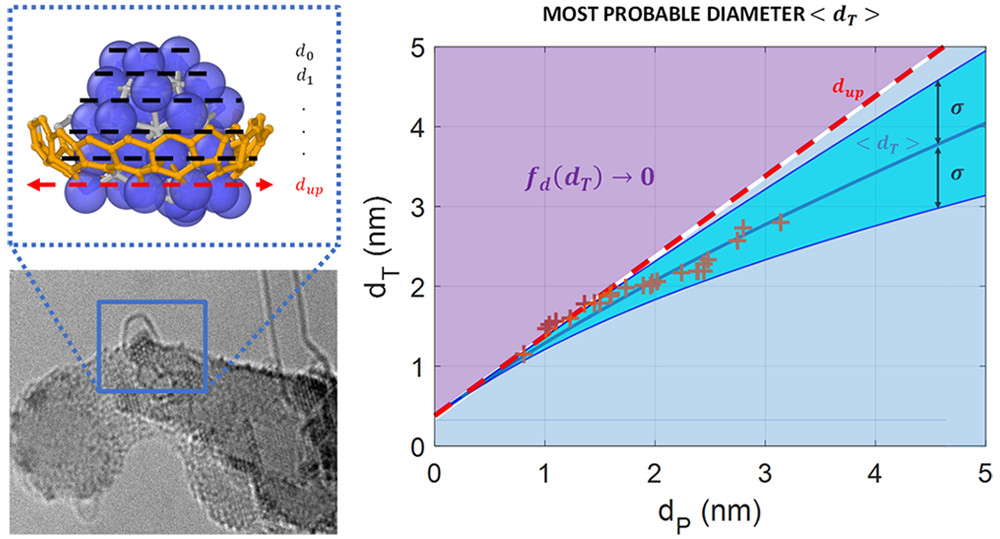
Mauricio C. Diaz, Hua Jiang, Esko Kauppinen, Renu Sharma and Perla B. Balbuena, “Can single-walled carbon nanotube diameter be defined by catalyst particle diameter?,” J. Phys. Chem. C, 123, 30305-30317, (2019).
The need of designing and controlling single-walled carbon nanotube (SWCNT) properties is a challenge in a growing nanomaterials-related industry. Recently, great progress has been made experimentally to selectively control SWCNT diameter and chirality. However, there is not yet a complete understanding of the synthesis process, and there is a lack of mathematical models that explain nucleation and diameter selectivity of stable carbon allotropes. Here, in situ analysis of chemical vapor deposition SWCNT synthesis confirms that the nanoparticle-to-nanotube diameter ratio varies with the catalyst particle size. It is found that the tube diameter is larger than that of the particle below a specific size (dc ≈ 2 nm) and above this value is smaller than particle diameters. To explain these observations, we develop a statistical mechanics based model that correlates possible energy states of a nascent tube with the catalyst particle size. This model incorporates the equilibrium distance between the nucleating SWCNT layer and the metal catalyst (e.g., Fe, Co, and Ni) evaluated with density functional theory (DFT) calculations. The theoretical analysis explains and predicts the observed correlation between tube and solid particle diameters during growth of supported SWCNTs. This work also brings together previous observations related to the stability condition for SWCNT nucleation. Tests of the model against various published data sets and our own experimental results show good agreement, making it a promising tool for evaluating SWCNT synthesis processes.
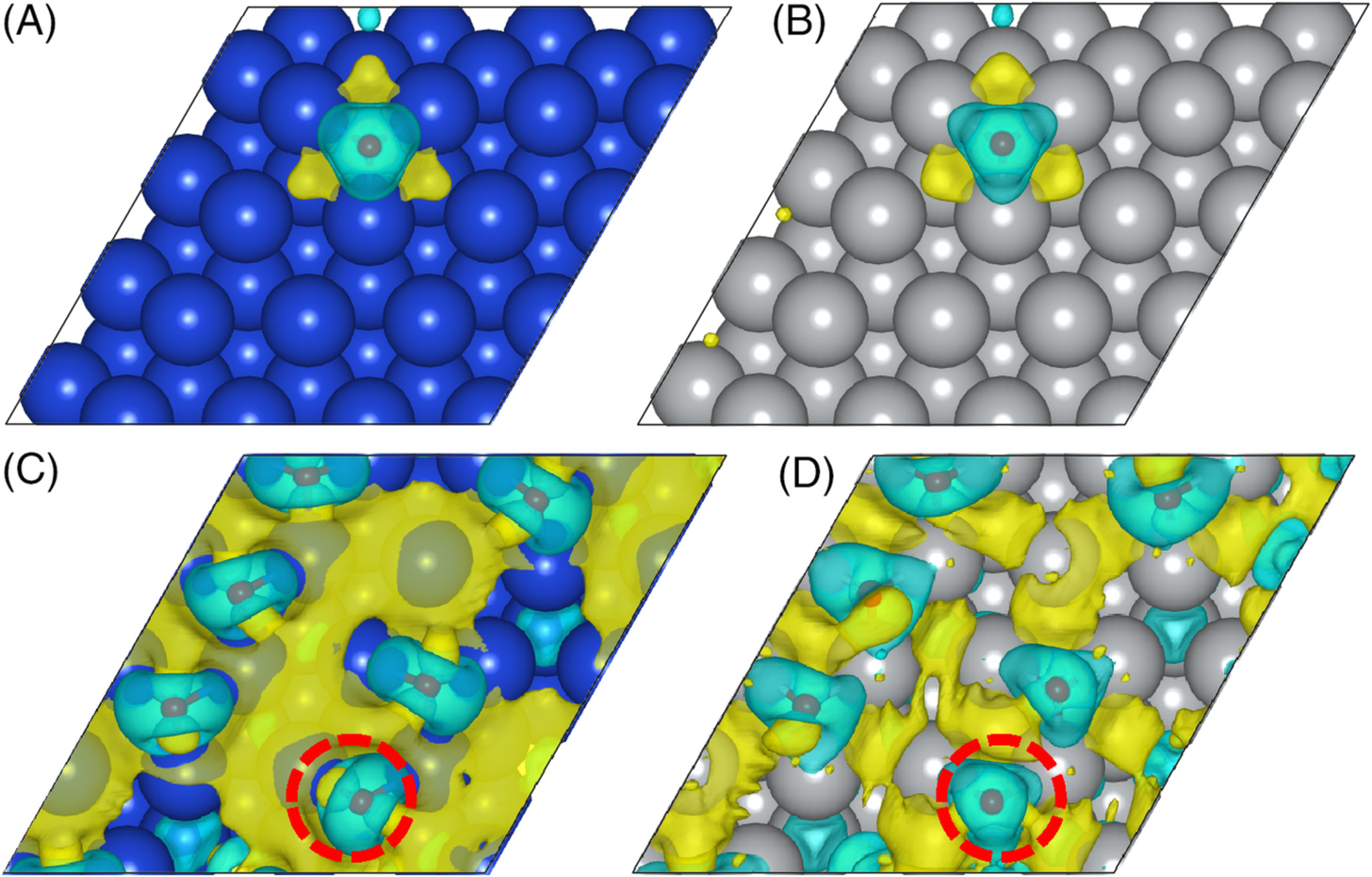
Behnaz Rahmani Didar and Perla B. Balbuena, “Methane Dehydrogenation on Cu and Ni Surfaces with Low and Moderate Oxygen Coverage,” Int. J. Quantum Chemistry, 2019; e26065.
First-principles density functional theory calculations are carried out to evaluate energy barriers and mechanisms for the dehydrogenation reactions of CH4 on clean and oxygen-covered surfaces of Cu (111) and Ni (111) with low and moderate oxygen coverage. In the presence of oxygen, two possible pathways have been evaluated. The more likely pathway, which is further analyzed, is that CH4 loses an H to the surface O. Results from this pathway agree with previous findings showing that oxygen promotes CH4 dissociation on Cu (111) and hinders that on Ni (111). In addition, our results show lower energy barriers on Cu with higher oxygen coverages up to 0.38 monolayer. However, such an increase in oxygen coverage did not show any favorable effect for CH4 dissociation on Ni (111). The findings are analyzed through electronic factors revealed by charge analysis and density of states.
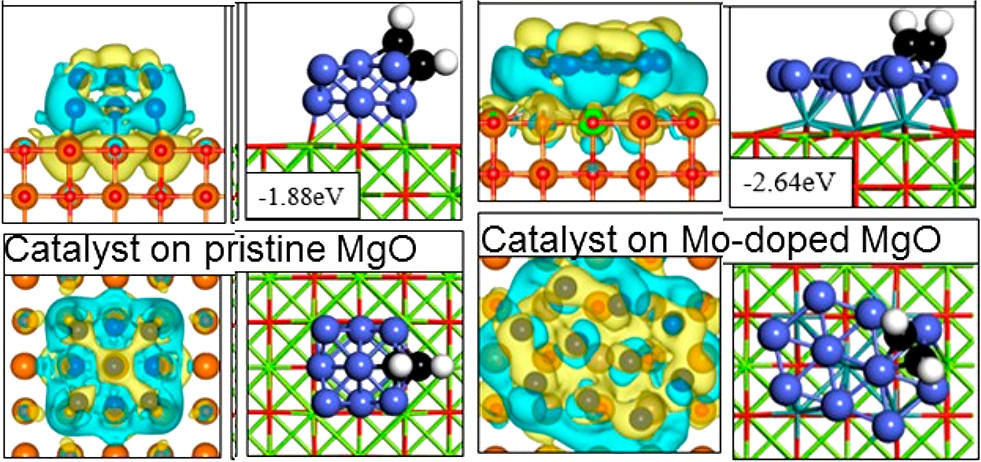
Behnaz Rahmani Didar and Perla B. Balbuena, “Reactivity of Cu and Co Nanoparticles Supported on Mo-doped MgO,” Ind.& Eng. Chem. Res., 58, 18213-18222, (2019).
First-principles density functional theory calculations are used to study the presence of Mo in the MgO support and its effect on the adsorption of Cu and Co nanocatalyst particles, as well as the mechanisms of C2H2 adsorption onto those nanoparticles. Furthermore, the initial steps of C2H2 dissociation on MgO-supported Cu and Co catalyst nanoparticles are investigated. Calculated formation energies suggest that the most likely locations of Mo-dopant atoms are on the top layer of the MgO support and under the overlying catalyst nanoparticles. The presence of Mo decreases the energy barriers for C2H2 dehydrogenation and CH–CH bond breakage on the Co nanoparticle catalyst, but it increases the energy barrier for CH–CH bond scission on the Cu nanoparticle catalyst. These energy barriers are considerably lower on Co compared to those on Cu nanoparticles. Overall, dehydrogenation is the most likely initial stage of the decomposition of C2H2 on both catalysts supported on Mo-doped MgO. The products of this dehydrogenation are found at nanoparticle corners/edges and along the interface with the support where charge transfer is greatest.
