
2-dimensional materials
Characterization
click images to enlarge:
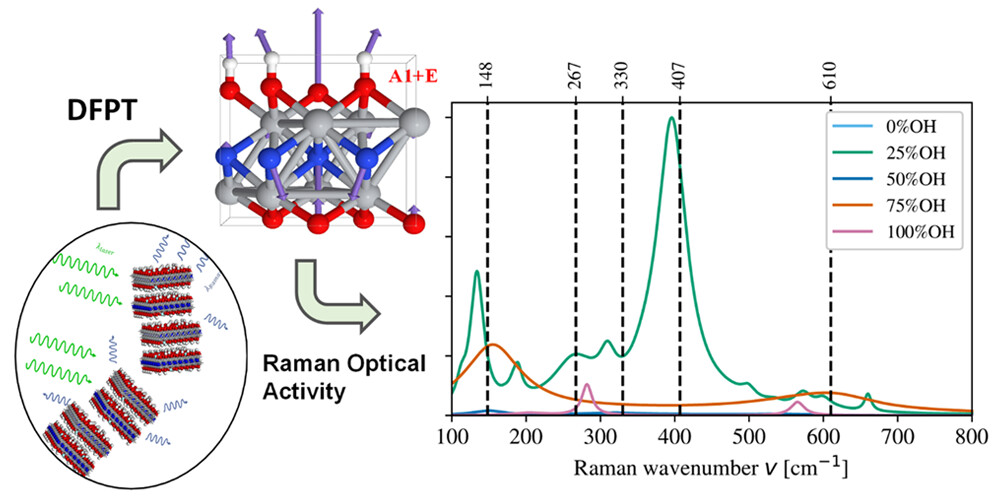
Hao-En Lai, Ray M. S. Yoo, Abdoulaye Djire, Perla B. Balbuena, “Investigation of the Vibrational Properties of 2D Titanium Nitride using DFT,” J. Phys. Chem. C, (2024).
Raman tensor analysis can be used to understand the surface chemistry of two-dimensional (2D) materials, which would enable the design of efficient MXene catalysts for electrochemical reactions. However, there are no reports of a detailed Raman tensor analysis on MXenes. In this work, we use density functional theory (DFT) Raman spectra calculations to provide insights into the surface chemistry of Ti2NTx MXene (Tx = −O– and/or −OH) by considering the influence of different Raman tensors on the spectra. We identify the Ti2NTx interlayer and surface termination as mostly −O– and −OH with small −F contributions based on optimized models, which influences MXene material reactivities. In this study we focus on the −OH termination group. Geometric tensor averages provide a better Raman spectra prediction for thick multilayer Ti2NTx MXene materials that match the experimental Raman spectra. In addition, we found the incident laser correction term to be essential for small wavenumber Raman intensities. Based on our Raman analysis, the surface termination coverages are either 25% −OH or 75% −OH. We found Raman intensities of 0%, 50%, and 100% −OH coverage to be negligible. Based on the simulated X-ray diffraction (XRD) results, the interlayer spacing is dictated by the interlayer −OH termination group. We also found that mismatched interlayer termination groups can restrict the interlayer distances. The present study provides insights into the role of the termination groups in stabilizing and influencing the structure of the MXene material, which, in turn, can be exploited to further enhance the application of MXenes in various energy conversion and storage systems.
Catalysis
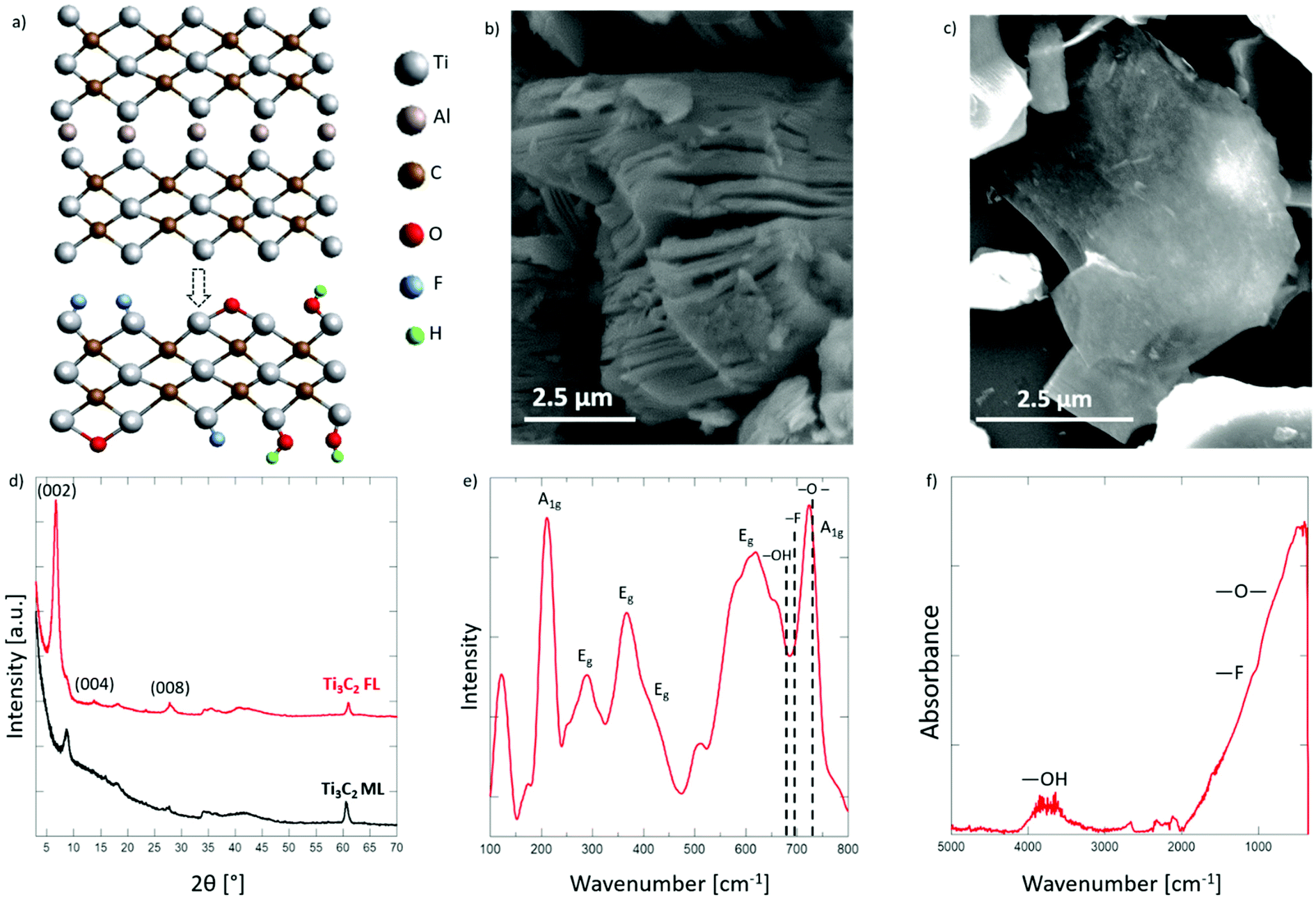
Denis Johnson, Hao-En Lai, Kyle Hansen, Perla B. Balbuena, and Abdoulaye Djire “Hydrogen evolution reaction mechanism on Ti3C2 MXene revealed by in situ/operando Raman spectroelectrochemistry”, Nanoscale, 14, 5068-5078, (2022).
MXenes have shown great promise as electrocatalysts for the hydrogen evolution reaction (HER), but their mechanism is still poorly understood. Currently, the benchmark Ti3C2 MXene suffers from a large overpotential. In order to reduce this overpotential, modifications must be made to the structure to increase the reaction rate of the H+/e− coupled transfer steps. These modifications heavily depend on understanding the HER mechanism. To remedy this, in situ/operando Raman spectroelectrochemistry combined with density functional theory (DFT) calculations are utilized to probe the HER mechanism of the Ti3C2 MXene catalyst in aqueous media. In acidic electrolytes, the –O– termination groups are protonated to form Ti–OH bonds, followed by protonation of the adjacent Ti site, leading to H2 formation. DFT calculations show that the large overpotential is due to the lack of an optimum balance between O and Ti sites. In neutral electrolytes, H2O reduction occurs on the surface and leads to surface protonation, followed by H2 formation. This results in an overcharging of the structure that leads to the observed large HER overpotential. This study provides new insights into the HER mechanisms of MXene catalysts and a pathway forward to design efficient and cost-effective catalysts for HER and related electrochemical energy conversion systems.
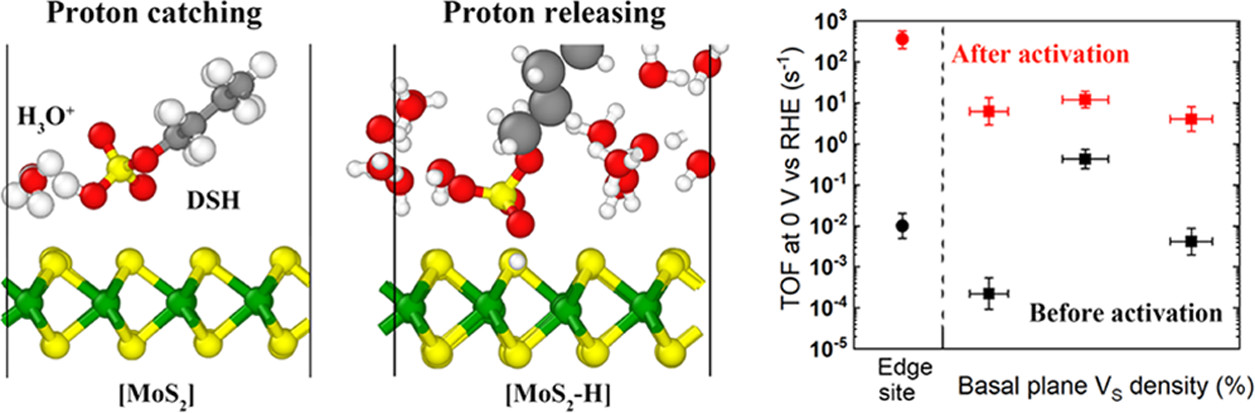
Xiangye Liu, Baichang Li, Fernando A. Soto, Xufan Li, Raymond R. Unocic, Perla B. Balbuena, Avetik R. Harutyunyan, James Hone, Daniel V. Esposito, “Enhancing Hydrogen Evolution Activity of Monolayer Molybdenum Disulfide via a Molecular Proton Mediator,” ACS Catalysis, 11, 19, 12159-12169 (2021).
The configuration and local environment of active sites in transition metal dichalcogenides can significantly alter their electrocatalytic activity toward the hydrogen evolution reaction (HER). Herein, we demonstrate that the HER activity of monolayer MoS2 electrocatalysts can be enhanced through the modulation of active sites by introducing a molecular mediator that alters the coverage of adsorbed protons. Sodium dodecyl sulfate (SDS) promotes the intrinsic HER activity of both terrace-based sulfur vacancies (VS) and edge sites during HER operation in an acidic environment, leading to increases in the turnover frequency (TOF) of both sites by up to 5 orders of magnitude. Simulations indicate that SDS facilitates proton adsorption by catching protons from hydronium ions and releasing them to VS, which reduces the energy barrier by creating a stair-case-like free energy profile. Our results highlight the ability to tailor the activity of electrocatalysts by synergistically combining proton transfer mediators with engineered active sites.
